
Translate this page into:
Diagnosis of posterior laryngeal cleft in a newborn with multiple congenital malformations
 , Dmitriev Andrey Vladimirovich1, Gudkov Roman Anatolyevich1, Terekhina Tatiana Anatolyevna1
, Dmitriev Andrey Vladimirovich1, Gudkov Roman Anatolyevich1, Terekhina Tatiana Anatolyevna1
*Corresponding author: Fedina Natalia Vasilyevna, Ryazan State Medical University, Ryazan, Russian Federation. k2ataka@mail.ru
-
Received: ,
Accepted: ,
How to cite this article: Vasilyevna FN, Igorevna PV, Vladimirovich DA, Anatolyevich GR, Anatolyevna TT. Diagnosis of posterior laryngeal cleft in a newborn with multiple congenital malformations. Karnataka Paediatr J 2023;38:140-3. doi: 10.25259/KPJ_55_2023
Abstract
The article describes a rare congenital malformation – laryngeal cleft in a newborn. Clinical manifestations are characterised by diverse non-specific symptoms in the form of dysphagia, cough, aspiration, and stridor. The severity of the condition is determined by the depth and degree of the defect and concomitant somatic and neurological pathology of the child. The article presents a clinical case of a Type 3 laryngeal cleft in a newborn with multiple congenital malformations and presents a diagnostic search for a cleft. At the age of 7 months, endoscopic suturing of the defect was performed, which allowed restoring enteral nutrition and normalizing the nutritional status of the child.
Keywords
Laryngeal cleft
Newborn
Diagnosis
Endoscopic correction

INTRODUCTION
The posterior cleft of the larynx is a rare congenital malformation diagnosed with a frequency of 1 in 10–20 thousand live births. The vice was first described by Richte in 1792. According to the classification of Benjamin and Inglis (1989), four types of laryngeal cleft are distinguished, supplemented, in 2006, by Sandu and Monnier, subtypes ‘a’ and ‘b’ in Types III and IV.[1,2]
Type I is a defect in the intercostal region extending to the level of the signet of the cricoid cartilage, Type II is a cleft partially extending into the signet of the cricoid cartilage, Type IIIa is a complete defect in the signet of the cricoid cartilage, Type IIIb is a defect extending partially to the trachea, Type IVa is a complete cleft or a complete tracheoesophageal fistula that can extend to tracheal bifurcation, Type IVb – a defect extending to the main bronchi. Laryngeal cleft is often combined with malformations of the gastrointestinal tract, heart, reproductive system, and craniofacial anomalies, but in an isolated form, prenatal diagnosis of this defect is not possible.[3]
From birth, choking, dysphagia, cough, aspiration pneumonia with episodes of bronchospasm, stridor, and hoarseness of voice are noted in the clinic. Diagnosis of cleft, when it is obvious, causes difficulties due to the variety of non-specific symptoms from not only the respiratory but also the digestive (regurgitation) system and the complexity of performing a number of diagnostic studies, including laryngoscopy, radiation methods, fibrogastroduodenoscopy, tracheobronchoscopy, especially if the child has another concomitant pathology. Despite the rarity of this defect, a possible laryngeal cleft should be suspected in any newborn with feeding problems and repeated aspiration, especially with respiratory insufficiency. The ‘gold’ standard of diagnostics is flexible video laryngoscopy, in which a doctor can study in detail the structure of the cartilage of the larynx and vocal cords from any angle and, if necessary, take photos and videos.
Medical and surgical treatment of laryngeal cleft is quite complicated and depends on the type and extent of the cleft, as well as the condition of the child and concomitant pathology. Therapy includes diet therapy, treatment of aspiration syndrome, tracheostomy, gastrostomy with fundoplication, as well as endoscopic suturing of the cleft using a cartilage endoprosthesis. When choosing a treatment tactic, some authors are in favour of performing surgery as early as possible to reduce aspiration. Others adhere to the tactics of palliative management of the patient with the intermediate establishment of gastrostomy and tracheostomy until stabilization of the condition and subsequent endoscopic suturing of the defect. We present our clinical observation of a child with multiple congenital malformations, including a cleft larynx.[4,5]
CASE REPORT
Boy B was born during the first pregnancy, the first delivery, by caesarean section at 39 weeks due to pelvic presentation.
Pregnancy was burdened by the threat of termination at 11 weeks and swelling of the feet at 18 weeks. During the entire pregnancy, the woman had asymptomatic bacteriuria (500 thousand Escherichia coli/mL) from 21 to 22 weeks – gestational pyelonephritis; at 23 weeks of gestation, ultrasound showed signs of incomplete intrauterine septum. In the third trimester of pregnancy, from 30 weeks, anaemia of the 1st degree was noted, in connection with which, the woman took iron preparations and, at 34 weeks – gestational thrombocytopenia.
Body weight at birth – 3560 g, body length – 57 cm, head circumference 34 cm, and chest circumference – 36 cm. The Apgar score is 8/9 points. When examining the child, multiple small developmental anomalies attracted attention: a wide, towering bridge of the nose, epicanthus, hypertelorism and a ‘Mongoloid’ eye incision, low-lying auricles, a short neck, a longitudinal defect of the frontal bone reaching the bridge of the nose, a stem form of hypospadias.
Eight hours after birth, the boy’s condition worsened; multiple regurgitations appeared, copious mucous discharge from the nose and oropharynx, tachypnoea and retraction of the compliant places of the chest, in connection with which the child was hospitalised in the department of pathology of newborns and premature infants of the GBU RO ‘CSTO named after N.V. Dmitrieva.’
On admission to the hospital, the child’s condition is regarded as severe due to respiratory manifestations and signs of respiratory insufficiency. There was nasolabial cyanosis, retraction of the compliant places of the chest, breath-hold 62/ min, SaO2 96%, with auscultation over the lungs, weakened breathing, and crepitating wheezing over the entire surface was heard. Systolic noise above the heart area, heart rate 148 per min, blood pressure on the right arm 68/38 mmHg, on the right leg 63/44 mmHg. At the age of 12.5 h from the moment of birth, an overview radiograph of the thoracic cavity revealed infiltration of the basal parts of the right lung. Additional examinations were also carried out: A neurosonogram without pathology. Echocardiogram: LLC, OAP 3.5 mm, atrial septum intact, aneurysmal protrusion with blood discharge from left to right 3.0 mm. Ultrasound examination of the abdominal cavity and kidneys revealed pyelectasis on the left up to 5 mm.
On a series of computed tomograms (CT) of the skull bones, there were no acute focal changes in the substance of the trunk, cerebellum, and large hemispheres of the brain; deformity of the bones of the facial part of the skull was revealed: the nasal septum was deflected to the right, a wide base of the nasal bones was noted, hypertelorism [Figure 1].

- (a and b) Computed tomography of the skull bones of patient B. Deviations of the nasal septum to the right, wide base of the nasal bones, hypertelorism.
Taking into account the persistent regurgitation syndrome, a computed tomogram of the chest organs was performed. Against the background of the picture of bilateral polysegmental pneumonia, along the posterior wall of the trachea in the upper third, there was a suspicion of the presence of a slit-like passage measuring 4 mm, which was regarded as probably a tracheoesophageal fistula [Figure 2].
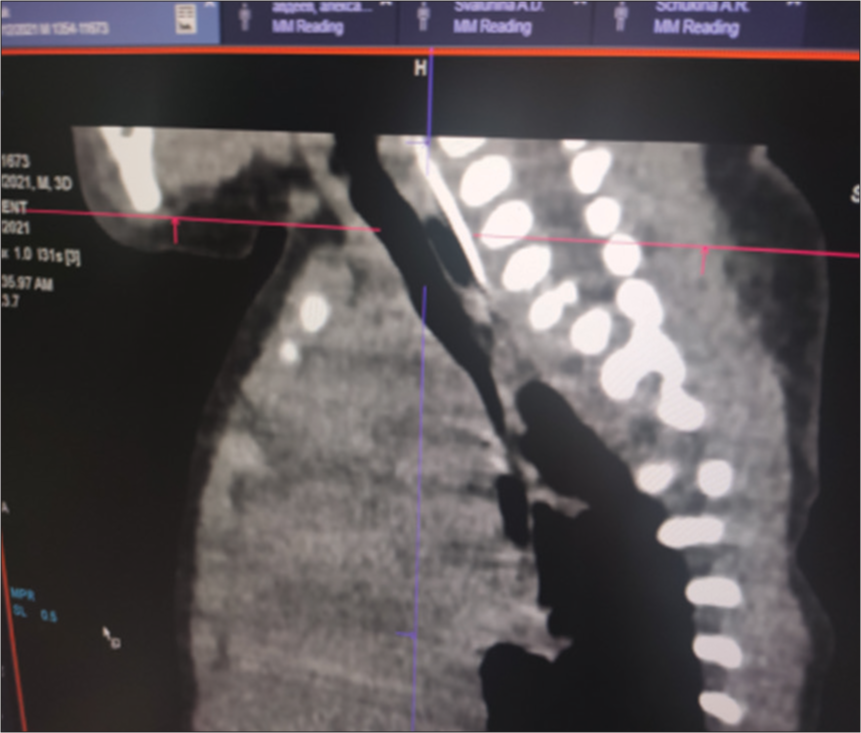
- Computed tomography of the chest organs: Slit-like passage in the upper third of the posterior wall of the trachea.
To confirm the latter, a fibrogastroduodenoscopic (FGDS) study was performed, which did not reveal the presence of a fistula. Fibrinous esophagitis and pronounced superficial gastritis were visualised. The child was examined by a geneticist, and frontonasal dysplasia (anomaly of the median cleft of the face) was diagnosed; Neurological and ophthalmological symptoms were not revealed.
Taking into account, the examination, the diagnosis was made: Congenital tracheoesophageal fistula. Congenital pneumonia, 0st. Fibrinous esophagitis. Pronounced superficial gastritis. Fronto-nasal dysplasia. Open oval window, atrial septal aneurysm, open ductus arteriosus. Hypospadias, stem form.
The child received treatment: partial parenteral nutrition, enteral feeding with expressed breast milk through a probe of 30.0 mL 8 times, antimicrobial (amoxicillin clavulanate, amikacin), antifungal (fluconazole) and inhalation (ipratropium bromide) therapy.
After stabilization of the condition, at the age of 10 days, the child was hospitalised in the department of intensive care and intensive care of newborns and infants of the Scientific Center for Children’s Health in Moscow of the Russian Federation. During bronchoscopy, splitting of hypoplasised cricoid cartilage was diagnosed with the formation of a laryngeal defect along the posterior wall, from the cricoid cartilage to the membranous part of the trachea, narrowing of the lumen of the left main and intermediate bronchi to 3 mm. The diagnosis was confirmed during FGDS, where from the pharyngeal-esophageal junction and distally, for about 3 cm, the esophagus communicates with the trachea due to a Type 3 laryngeal cleft [Figure 3].
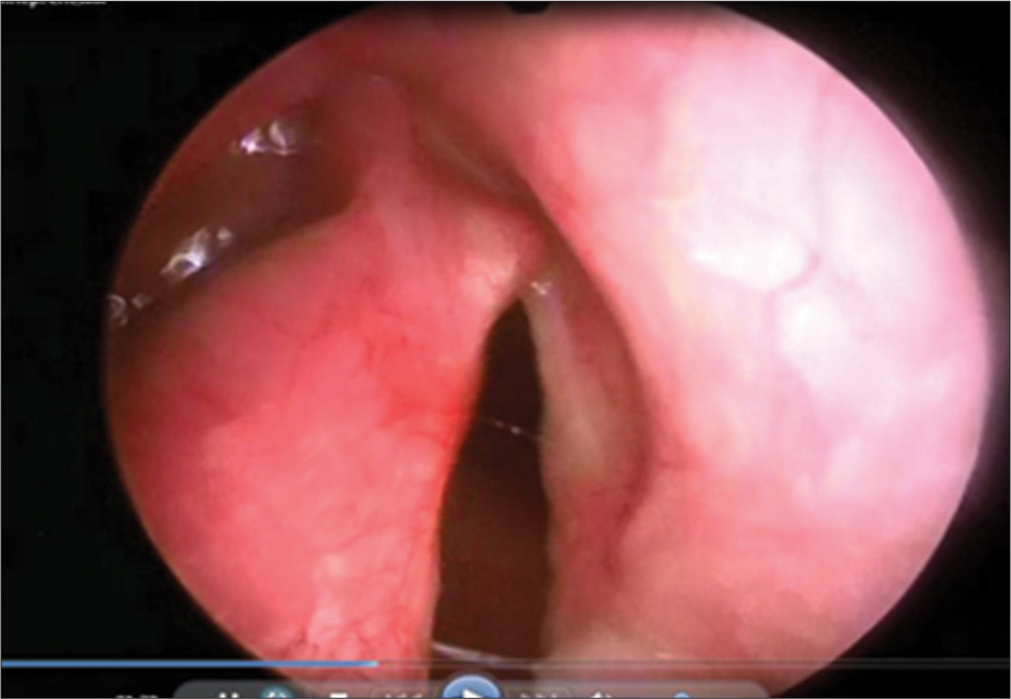
- Fibrogastroduodenoscopy: Laryngeal cleft of the 3rd degree.
Thus, after a more in-depth examination, the diagnosis was made: Laryngeal cleft Type 3, narrowing of the lumen of the left main bronchus and intermediate bronchi.
Due to the presence of hypospadias, during repeated ultrasound examinations of the kidneys, a left-sided megaureter was diagnosed, and vesicoureteral reflux was also suspected [Figure 4]. After X-ray contrast examination (excretory urography and cystography), a picture of bilateral vesicoureteral reflux of II and III degrees was revealed [Figure 5].
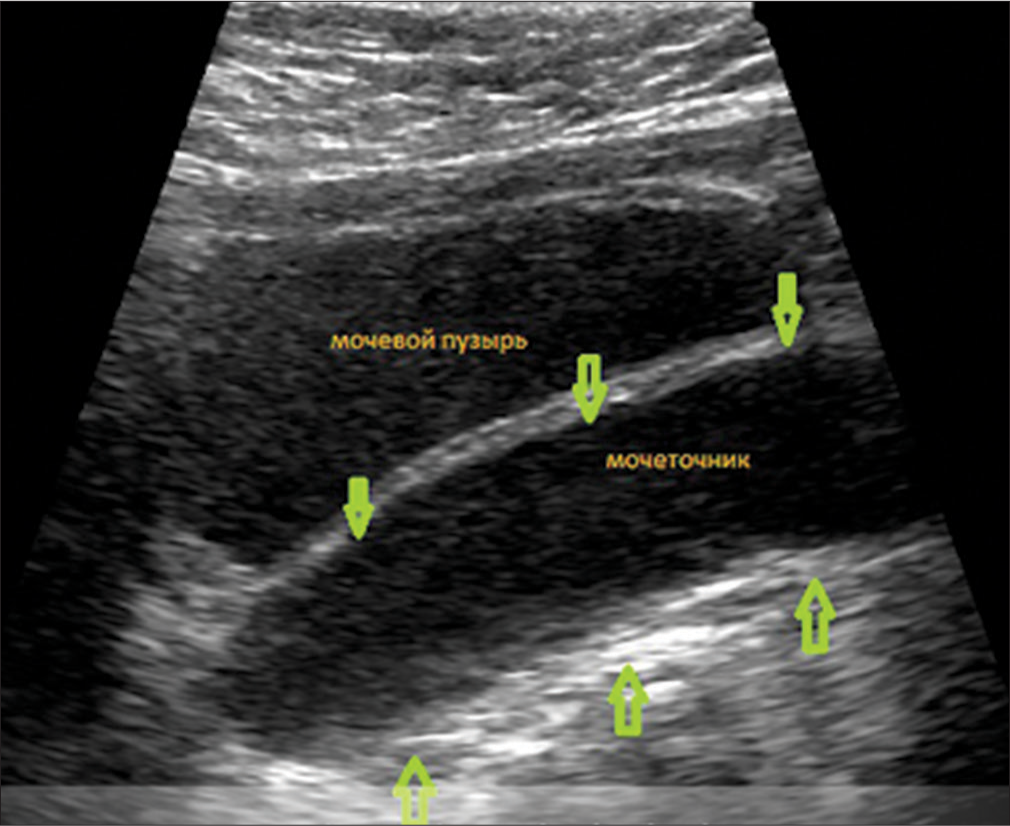
- Ultrasound of the kidneys and bladder: Signs of a megaureter on the left, thickening of the walls of the ureter. The green arrows indicate the thickened walls of the ureter.
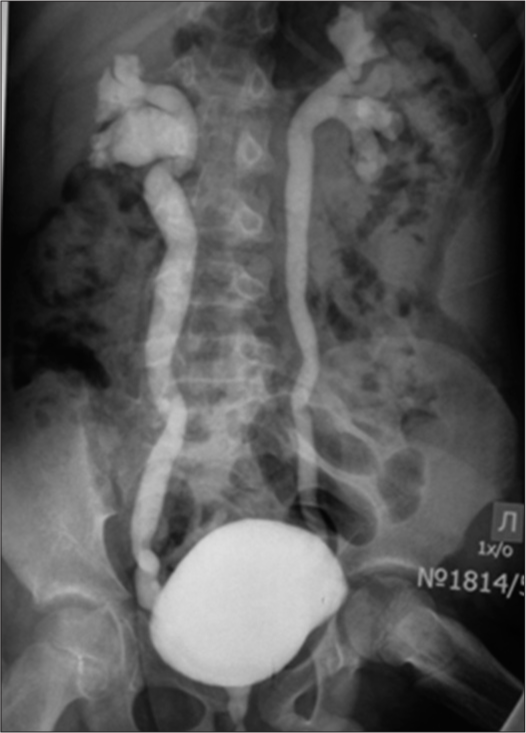
- Mictional cystogram: A picture of bilateral vesicoureteral reflux of II and III degrees, megaureter.
Based on the clinical and additional examination data, the final clinical diagnosis was formulated.
Main: Multiple malformations: Laryngeal cleft Type 3a. Narrowing of the lumen of the left main and intermediate bronchi. Hernia of the esophageal orifice of the diaphragm. Double-sided megaureter. PMR of the III degree from two sides. Stem hypospadias. Frontonasal dysplasia. Concomitant: Congenital aspiration pneumonia, respiratory failure of the 1st degree. Fibrinous esophagitis. Pronounced superficial gastritis.
The child underwent endoscopic gastro fundoplication, gastrostomy, and ureteral endoplasty on both sides. After discharge from the hospital, the child is fed through a gastrostomy with breast milk. At the age of 2.5 months, the boy suffered a respiratory syncytial viral infection twice, at 4 and 5.5 months, obstructive bronchitis. At six months, the patient had a weight of 6700 g (body mass index = 14.5), which corresponds to protein-energy deficiency of the 1st degree, neuropsychiatric development without deficiency. At the age of 7 months, a radical correction of the defect was performed: Endoscopic suturing of the cleft using its cartilage as an endoprosthesis.
DISCUSSION
The described clinical case demonstrates the difficulty of diagnosing a cleft larynx in a newborn. Attention is drawn to the early appearance of such non-specific symptoms as dysphagia, cough, aspiration pneumonia, and stridor. The primary diagnostic measures carried out did not reveal data for the laryngeal cleft, which is probably due to the complexity of the invasive interventions carried out, as well as the accompanying multiple malformations, which also determined the severity of the patient’s condition. The absence of data for tracheoesophageal fistula according to primary FGDS did not entail further diagnostic interventions, including bronchoscopy. Prolonged preservation of symptoms (regurgitation, foamy vomiting) against the background of diet therapy with some stabilization of the patient’s condition allowed us to continue the diagnostic search and detect a deep defect in the development of the larynx. The diagnosis was confirmed by the results of repeated FGDS and bronchoscopy. Radical correction of the defect in the form of endoscopic suturing of the cleft, using its cartilage as an endoprosthesis, was carried out only at the age of 7 months, after a period with the installation of a gastro and tracheostomy to the patient.
CONCLUSION
Congenital anomalies of the structure of the larynx can manifest from the birth of a child in the form of respiratory disorders, phonation, and the work of the upper gastrointestinal tract. The severity of clinical manifestations is determined not only by the type of cleft but also by the somatic and neurological status of the patient. Endoscopic methods of laryngeal visualization play a leading role in the diagnosis. Further management tactics of the infant, depending on the extent of the laryngeal cleft, may consist of several steps of endosurgical treatment, including, at the first stage, in patients with type 2–4 cleft, the imposition of a tracheostomy and gastrostomy. In some cases, it is possible to use not only endoscopic techniques but also the open-access method. Measures aimed at disconnecting the respiratory and digestive pathways are carried out for all types of this anomaly. The effectiveness of the treatment depends on the severity of concomitant congenital malformations in each patient. Endoscopic surgical treatment makes it possible to radically eliminate the existing defect and restore the enteral nutrition of the patient.
Ethical approval
The research/study complied with the Helsinki declaration of 1964.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript, and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Endoscopic laryngotracheal cleft repair without tracheotomy or intubation. Laryngoscope. 2006;116:630-4.
- [CrossRef] [PubMed] [Google Scholar]
- Laryngeal cleft: Evaluation and management. Int J Pediatr Otorhinolaryngol. 2014;78:905-11.
- [CrossRef] [PubMed] [Google Scholar]
- Laryngeal cleft: A literature review. Am J Otolaryngol. 2021;42:103072.
- [CrossRef] [PubMed] [Google Scholar]
- Current management of Type III and IV laryngotracheoesophageal clefts: The case for a revised cleft classification. Curr Opin Otolaryngol Head Neck Surg. 2020;28:435-42.
- [CrossRef] [PubMed] [Google Scholar]




