
Translate this page into:
A rare case of genetically proven neurofibromatosis 1 with Charcot-Marie-Tooth disease, type 4C

*Corresponding author: Vykuntaraju K. Gowda, Department of Paediatric Neurology, Indira Gandhi Institute of Child Health, Bengaluru, Karnataka, India. drknvraju08@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Gowda VK, Challa VR, Srinivasan VM. A rare case of genetically proven neurofibromatosis 1 with Charcot-Marie-Tooth disease, type 4C. Karnataka Paediatr J. doi: 10.25259/KPJ_15_2025
Abstract
With sophisticated technological improvements, genetic testing has become easily available. Different genetic conditions can coexist in a single individual, and it is important to identify them to manage the child confidently and effectively and to prognosticate. Here, we present a 9-year-old male child, with predominant motor delay with regression of attained motor milestones. Facial dysmorphism with strabismus, multiple café-au-lait macules with axillary freckles, flat foot with wasting and kyphoscoliosis were noted. Neurological examination revealed decreased tone, distal power of 2/5 and sluggish deep tendon reflexes, while other systems were normal. The magnetic resonance imaging brain, orbits and spine was normal. Nerve conduction studies (NCS) revealed demyelinating polyneuropathy. Genetic analysis revealed NF1 and SH3TC2 genes causing NF-1 with Charcot-Marie-Tooth type 4C with phenotypic correlation. To conclude, a thorough clinical examination, NCS and exome sequencing were used to definitively diagnose two distinct disorders in a single child after a clinical diagnosis of NF1 with coexisting peripheral neuropathy created a diagnostic conundrum.
Keywords
Coexistent conditions
Genetics
Hereditary motor and sensory neuropathy
Neurofibromatosis type 1

INTRODUCTION
An autosomal dominant condition known as neurofibromatosis type 1 (NF1; OMIM #162220) is characterized by a variety of tumor forms, primarily neurofibromas. NF1 is diagnosed based upon the presence of 2 or more of the following criteria: multiple café-au-lait macules, 2 or more neurofibromas (or 1 plexiform neurofibroma), a family history of NF1, axillary/inguinal freckling, optic nerve glioma, 2 or more Lisch nodules, or bone dysplasia.[1-3] Extreme foot deformities (pes cavus, pes planus, or pes valgus) and severe spine deformities (scoliosis or kyphoscoliosis) are hallmarks of SH3TC2-related hereditary motor and sensory neuropathy (SH3TC2-HMSN, OMIM #601596) a demyelinating neuropathy inherited in the autosomal recessive state, that usually manifests in the first decade of life or early adolescence. Additional observations may include breathing issues, cranial nerve involvement (most frequently shown as tongue involvement), facial weakness or paralysis, hearing loss, or dysarthria.[4] Here we report a rare and, to our knowledge, very first case of genetically proven NF-1 with CMT type 4C in an Indian child.
CASE REPORT
A 9-year-old male child, second born to non-consanguineously married parents, with no significant perinatal history, presented with mild initial global developmental delay followed by progressive difficulty in walking for 2 years. The child noticed distal muscle weakness of bilateral lower limbs in the form of difficulty in wearing slippers for 2 years, followed by upper limb distal muscle weakness in the form of difficulty in buttoning, unbuttoning and change in handwriting. History of distal limb muscle wasting without any cranial nerve involvement and bowel and bladder incontinence. General examination revealed a weight of 18 kg (−4.15 Z), head circumference of 50 cm (−2.41 Z), dysmorphism with strabismus and multiple café-au-lait macules (CALMS) with axillary freckles with similar multiple CALMS in mother and other sibling-sister: Mild facial: Asymmetry, flat foot with wasting and kyphoscoliosis [Figure 1a-e]. Neurological examination revealed decreased tone, power of distal 2/5 in distal 3/5 in a proximal group of muscles and sluggish deep tendon reflexes while other systems were normal. The magnetic resonance imaging of the brain, orbits and spine were normal. Nerve conduction studies revealed demyelinating polyneuropathy. Genetic testing revealed variations in two genes, SH3TC2 and NF1 genes. In the SH3TC2 gene, we identified two heterozygous variants in trans; first one is a known nonsense variant, c.2710C>T, p.(Arg904Ter) in exon-11 classified as pathogenic (PVS1, PM2, PP5) as per ACMG classification, second one is a novel frameshift deletion c.1564delC, p.(His522Thrfs*2) in exon-11 classified as likely pathogenic (PVS1, PM2). In NF1, we identified a known splice site variant c.288+1G>A in Intron-3 classified as pathogenic (PVS1, PM2 and PP5).
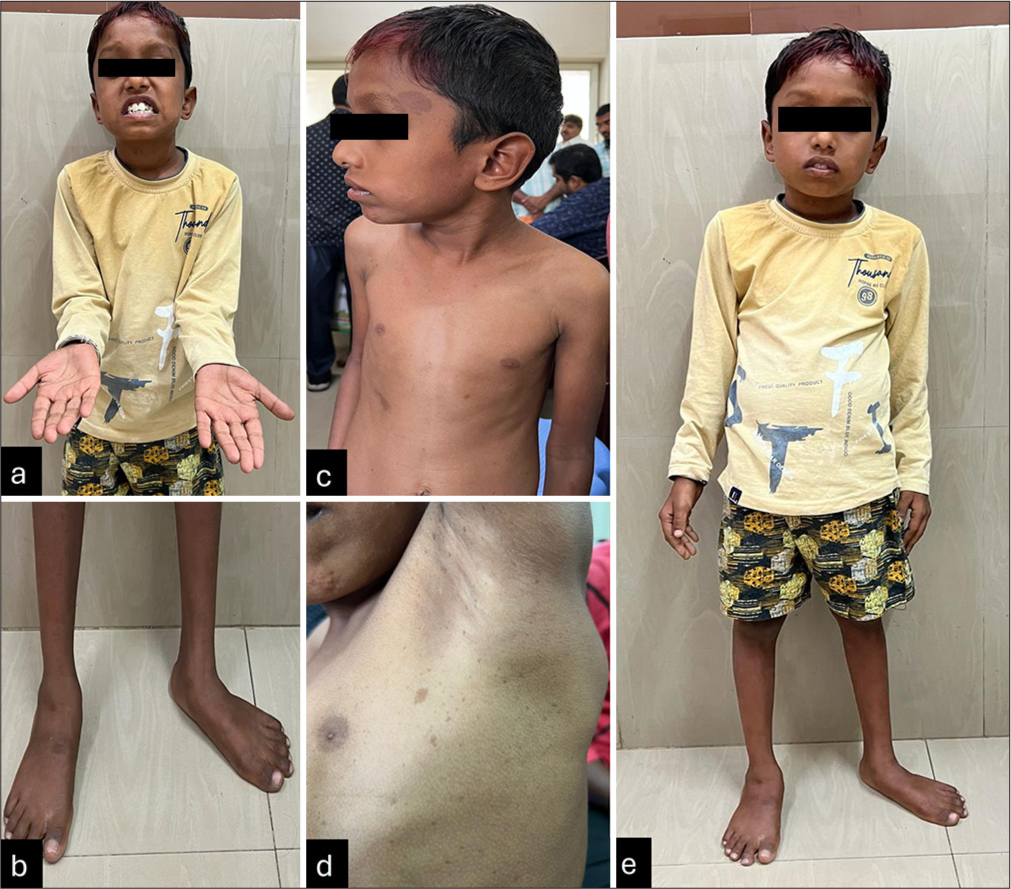
- (a and b) Show distal wasting with flat feet, (c) shows multiple café-au-lait macules, (d) shows axillary freckling, (e) shows a clinical photograph of the child with flat feet.
DISCUSSION
NF1 is the more common of the two conditions. CMT symptoms usually appear in teens and young adults. Most newborns with NF1 do not have symptoms, but some have curved lower leg bones. By their first birthday, most children with NF1 have several CALMS. On chromosome 17, it is brought on by mutations that cause the NF1 gene to stop functioning. Although some mutations are difficult to identify due to the huge size of the NF1 gene, 95% of individuals with NF1 can have NF1 mutations found by the most thorough genetic testing currently available.
NF1 and CMT to co-exist are rare but still possible. Both NF1 and CMT are genetic conditions that affect the peripheral nervous system. By describing the above case, more than one disorder is possible if clinical features are not able to explain a single condition, as diagnosis of both conditions is important for treatment, monitoring of disease and genetic counselling.
CONCLUSION
A clinical diagnosis of NF1 with coexisting peripheral neuropathy can cause a diagnostic dilemma; detailed clinical examination, nerve condition studies and exome sequencing helped in the definitive diagnosis of two different disorders in a single child.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that they have used artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript or image creations.
Financial support and sponsorship: Nil.
References
- Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124-33.
- [CrossRef] [PubMed] [Google Scholar]
- SH3TC2-Related hereditary motor and sensory neuropathy In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993.
- [Google Scholar]
- Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541-55.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10:508-15.
- [CrossRef] [PubMed] [Google Scholar]




