
Translate this page into:
A rare case of Klippel–Trenaunay syndrome in a neonate
*Corresponding author: Sukrutha Surandran, Senior resident, Department of Pediatrics, Indira Gandhi Institute of Child Health, Bengaluru, Karnataka, India. sukrutha.world94@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Surandran S. A rare case of Klippel–Trenaunay syndrome in a neonate. Karnataka Paediatr J 2023;38:131-4. doi: 10.25259/ KPJ_7_2023
Abstract
Klippel–Trenaunay syndrome (KTS) is a rare syndrome of congenital capillary-venous vascular malformation associated with altered limb bulk and/or length. It is characterised by capillary nevus, varicosities and hypertrophy of bones and tissues of the affected limb. These three features constitute the primary diagnostic criteria of this syndrome. Although the cause of this disorder is unknown, it has a sporadic occurrence with some reports mentioning autosomal dominant transmission as well. The diagnosis of this embryological malformation is mainly clinical along with supportive evidence from other imaging modalities. In this article, we report a 5-dayold male baby who had phenotypic features suggestive of KTS and was later confirmed by magnetic resonance imaging.
Keywords
Bony hypertrophy
Klippel–Trenaunay syndrome
Soft-tissue hypertrophy
Varicosities

INTRODUCTION
Klippel–Trenaunay syndrome (KTS) is a rare congenital disorder with an incidence of 3–5/1,00,000. It is characterised by the clinical triad constituting vascular malformation of capillaries (port wine stain), venous varicosities and sometimes lymphatic types, bone and soft-tissue hypertrophy.[1] The clinical manifestation can vary from classical triad to varied presentation including the development of potentially life-threatening complications, such as pulmonary embolism, deep vein thrombosis and massive gastrointestinal (GI) bleeding. The anomaly if present is present at birth and usually involves the lower limbs as well as a portion of the trunk, face, upper limb or head. Although the clinical presentation is typical, radiological investigations help in confirmation. Diagnosis is usually made when two of the three features are present. Management includes careful diagnosis, prevention and treatment of complications.[1,2]
Herein, we describe a 5-day-old male baby who presented to us for evaluation of the lower limbs swelling and had radiological changes described in association with KTS.
CASE REPORT
A 5-day-old term boy neonate, appropriate for gestational age born second to a 22-year-old mother with 2nd-degree consanguinity, by normal vaginal delivery and said to have cried immediately after birth with good Apgar scores and birth weight of 2.90 g was referred to our institution for evaluation of the lower limb swelling [Figure 1a and b].

- (a and b) Lower limb swelling.
On examination baby had normal vitals of heart rate - 130 bpm and well felt peripheral pulses, respiratory rate - 34 cpm, warm peripheries, blood pressure - 70/40 and head-to-toe examination revealed diffuse hypertrophy of left thigh, leg and foot, hypertrophy of all the digits with length discrepancy of 1 cm and undescended testes on the left side, there were no facial dysmorphisms, no port wine stain, no neurocutaneous markings, anthropometry was within acceptable range with weight 2.9 kg, head circumference of 35 cm and length 49 cm. There was no organomegaly on per abdomen examination and respiratory, cardiovascular and neurological examination was normal. A provisional diagnosis of congenital venous malformation or lymphatic malformation, arteriovenous malformation (AVM) was considered and evaluated.
Routine haematological examination showed haemoglobin of 12.7g, white blood cells - 12300, platelets - 2.4 lakhs, C-reactive protein was negative (sepsis screen-negative), renal, liver and thyroid function tests were within normal limits. X-ray pelvis with hip and left thigh showed soft-tissue hypertrophy [Figure 2].
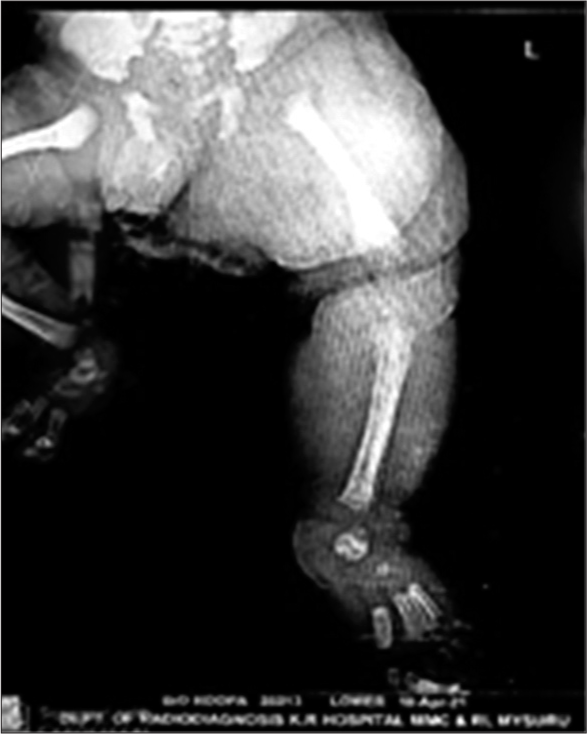
- X-ray findings - soft tissue hypertrophy.
Ultrasonography showed bulky thigh and calf muscles with mild loss of fibrillar pattern, colour Doppler showed dilated superficial veins, no evidence of venous thrombosis noted in either of deep or superficial veins, no obvious abnormalities of the deep venous system were seen and no collection in joint cavity [Figure 3a-c].
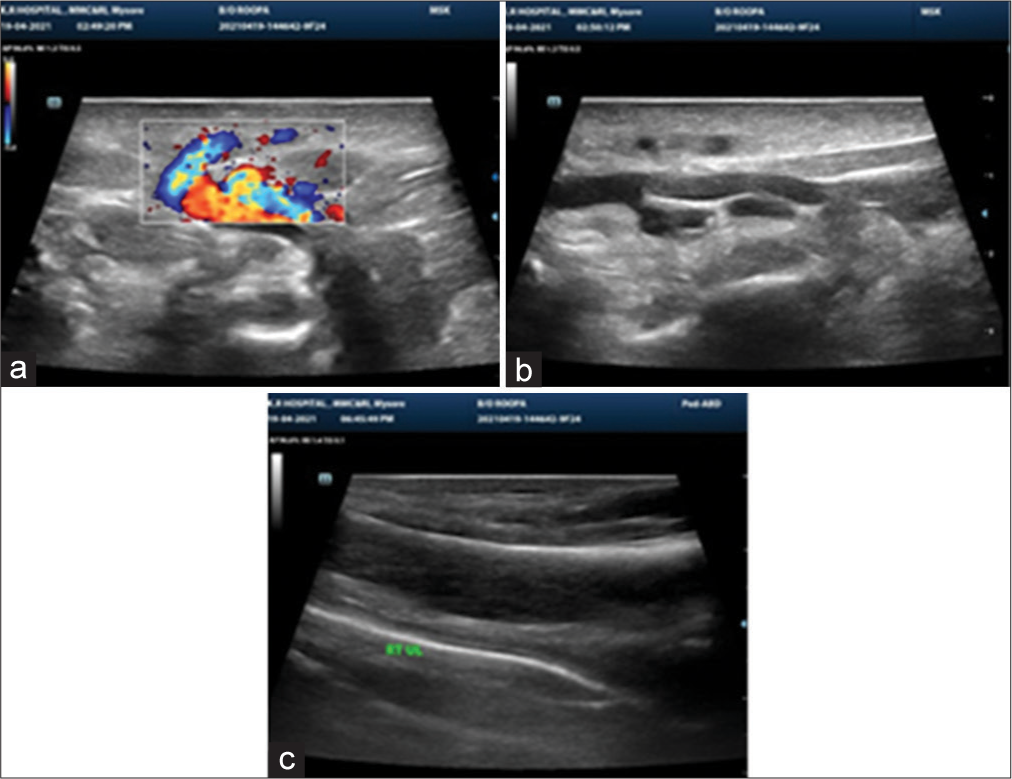
- (a-c) USG thigh and calf showing bulky soft tissues with loss of fibrillar pattern.
Magnetic resonance imaging (MRI) showed soft-tissue hypertrophy of the left lower limb, dilated superficial venous system on the left side, prominent left lower limb arterial system and no abnormal AVM [Figures 4a and b, 5a and b]. All these features concern congenital vascular malformation most probably Klippel–Trenaunay syndrome or less likely park–weber syndrome.
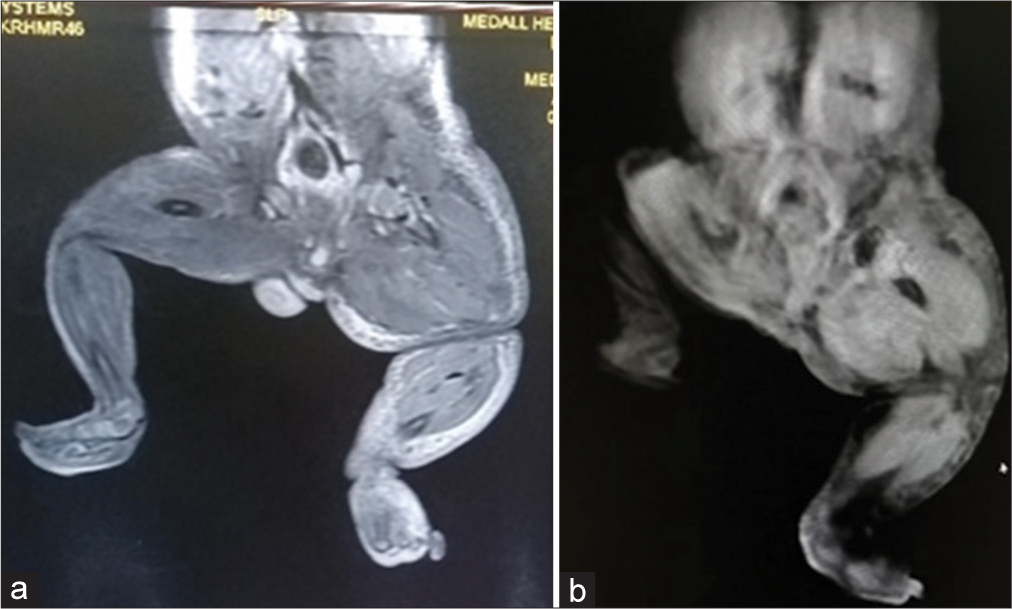
- (a and b) MRI showing soft tissue hypertrophy of the left lower limb.
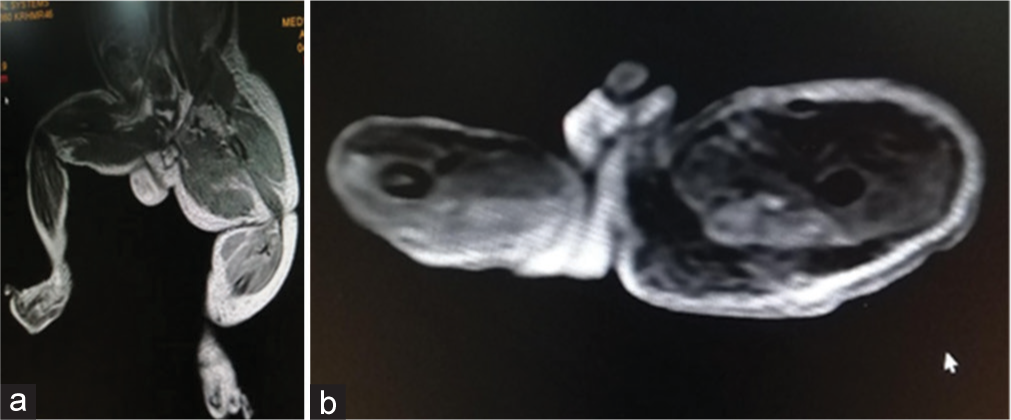
- (a and b) - MRI showing dilated superficial venous system with prominant arterial system of the left lower limb.
As the child did not have any complications, parents were reassured and informed of the possible complications including cellulitis, pain, limb length discrepancy, bleeding manifestations and coagulopathy and were advised to come for regular follow-up for limb length monitoring and to look for complications by imaging including magnetic resonance angiography (MRA), venography, biopsy and genomic studies if required. Treatment options for leg length adjustments (inserting lift in shoes), infection and pain control, sclerotherapy and endovascular laser for capillary malformations, anticoagulants, the role of newer therapies mTOR inhibitors -sirolimus, PIK3CA inhibitors (alpelisib) and various surgical options (epiphysiodesis, staged contour resection) were explained to the parents.
DISCUSSION
KTS – a rare congenital vascular disorder was first described by two French physicians, Klippel and Trenaunay in 1900. Most cases are sporadic with an equal proportion of males and females being affected, with no racial predilection. It usually manifests at birth or during childhood with varied presentation. KTS is characterised by a triad of capillary malformation, venous or lymphatic varicosity with soft tissue and bony hypertrophy. This condition was earlier confused with Park Weber syndrome which was later found to be entirely different.[2]
The aetiology of KTS remains unknown. KTS is most commonly a sporadic event. Several theories had been proposed earlier such as Servelle’s theory many of which was found obsolete later. Some studied described genetic association where it is found to be caused by a mutation in the PIK3CA gene. This gene is needed for the production of p110alpha protein, a subunit of phosphatidylinositol-kinase (PI3K) which plays a role in chemical signalling that is important for many cell proliferation, migration and survival important for tissue development. Mutation results in abnormally active PI3K which allows cells to grow and divide continuously-leading to abnormal growth of bones, soft tissues and blood vessels. However, since this mutation was not found in everyone with KTS, other unidentified genes may also cause this condition.[2]
KTS has a myriad of clinical presentations from mild or incomplete forms of port wine stains and few varicose veins causing only cosmetic deformity to severe disability associated with massive limb overgrowths, chronic pain syndrome, infections of skin and joints, thromboembolism and life-threatening bleeding from venous malformations most commonly pelvic or rectal.[3]
Capillary malformations are the most common cutaneous manifestation of KTS. Vascular changes found in KTS are congenital where vessels are improperly formed and remodelled. Typically involves the limb, although any part of the body can be affected. The lower limb is the most common site of malformations, seen in approximately 95% of patients. Hypertrophy is the most variable of the three classic features. Extremities can be enlarged either in the form of bone elongation, circumferential bone hypertrophy, or both, often presenting as limb length discrepancy, not significant enough to necessitate orthopaedic intervention. Varicose veins are present in the majority of patients, this can occur in both the superficial and deep venous systems.[3-5]
These patients can have additional malformations other than the triad. Craniofacial asymmetric facial hypertrophy, micro or macrocephaly, intracranial calcifications, eye abnormalities-glaucoma, cataract, skeletal-syndactyly, polydactyly, oligodactyly, macrodactyly, disproportionate growth of digits, skin-hyperpigmentation nevi, ulcers, vesicles, telangiectasia and viscera-visceromegaly, aberrant blood vessels, others seizures, genital enlargement, coagulation defects, oesophageal varices, hematuria and hematochezia. These variations in symptoms suggest that KTS is genetically heterogeneous and affected by multiple factors.[4-6]
Complications are related to vascular pathologic processes. Stasis dermatitis, thrombophlebitis, cellulitis and serious complications such as thrombosis, coagulopathy, pulmonary embolism, congestive cardiac failure and bleeding from abnormal vessels in the gut, kidney and genitalia can occur. Rectal and bladder haemorrhages are serious complications in pelvic vascular malformation. Bleeding is the earliest and most common symptom in GI tract involvement, most common sites being the distal colon and rectum.[3]
Diagnosis of KTS is mainly clinical. Investigations in KTS focus on the evaluation of the type, extent and severity of malformation and confirm the absence of any clinically significant arteriovenous shunting. Imaging plays an important role in the diagnosis and evaluation of KTS. In plain radiography, phleboliths are diagnostic of venous malformation in young patients. Barium studies show luminal narrowing of the affected small and large bowel with scalloped mucosal outline caused by the presence of varicosities or sub-mucosal vascular malformation. Ultrasonography identifies abnormal veins and varicosities. Contrast venography and computed tomography scans are required to evaluate a deep venous system and collateral formation. MRI is performed on different bone, fat and muscle hypertrophy, to assess the extent of soft-tissue involvement and vascular malformation.[2,3]
In our case, Parkes–Weber syndrome (PWS) can be considered a close differential diagnosis. PWS is characterised by overgrowth of the affected limb combined with arteriovenous fistula or shunt. It is difficult to distinguish both syndromes in routine clinical practice and they are easily misdiagnosed. Doppler ultrasound and MRA studies aid in reaching the proper diagnosis and are helpful to radiologically differentiate between these two conditions.[3]
KTS treatment involves a multi-disciplinary approach, consultation with a paediatrician, dermatologist, interventional radiologist, plastic surgeon, orthopaedic surgeon, vascular surgeon, haematology, urology and gastroenterology is required. No definitive treatment is identified. Treatment is aimed at reducing the symptoms and preventing complications. Active intervention is done only when there is a localised lesion or serious complication such as bleeding or cardiac failure is present. Conservative management includes compression garments, lymphatic massage, physical therapy and physical activity. Other treatment options include sclerotherapy, laser therapy, thermal ablation and surgery. Treatment of GI and genitourinary vascular malformations depends on the severity of blood loss and includes resection of the diseased bowel, and partial cystectomy. If leg length discrepancy exceeds 2 cm at the time of skeletal maturity epiphysiodesis can be done.[4,6,7]
Patients need monitoring at least annually and more frequently if clinical symptoms are present. If there is a progression of disease, imaging studies should be done and proper intervention carried out.
CONCLUSION
We are reporting here a common presentation of an uncommon syndrome that has widely variable clinical presentations from mild symptoms to fatal complications like internal organ bleeds. Increasing awareness of KTS as a diagnosis is vital to avoid misdiagnosis or delay in recognising this syndrome.
Ethical approval
The research/study complied with the Helsinki declaration of 1964.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The author confirms that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- National org for rare disorder (NORD) 2023. Available from: https://raredisorder.org?rare-disease?pik3ca-relatedovergrowthspectrum [Last accessed on 2023 Jun 20]
- [Google Scholar]
- Medline plus genetics. 2021. Philadelphia, PA: Medlineplus; Available from: https://medlineplus.gov/genetics/condition/klippeltrenaunaysyndrome/ [Last accessed on 2023 Jun 20]
- [Google Scholar]
- KlippelTrenaunay and Parkes-Weber syndromes: Two case reports. J Vasc Bras. 2017;16:320-4.
- [CrossRef] [PubMed] [Google Scholar]
- Klippel-Trénaunay syndrome-a very rare and interesting syndrome. Clin Med Insights Circ Respir Pulm Med. 2015;9:1-4.
- [CrossRef] [PubMed] [Google Scholar]
- Radiological aspect of Klippel-Trénaunay syndrome: A case series with review of literature. Curr Med Sci. 2018;38:925-31.
- [CrossRef] [PubMed] [Google Scholar]
- Foot or hand malformations related to deep venous system anomalies of the lower limb in KlippelTrénaunay syndrome. J Am Acad Dermatol. 2009;61:621-8.
- [CrossRef] [PubMed] [Google Scholar]




