
Translate this page into:
An unusual presentation of biotinidase deficiency in infant: High anion gap metabolic acidosis
-
Received: ,
Accepted: ,
How to cite this article: Wali PS, Tauro P, Hegde P, Khan HU, Jaidev MD. An unusual presentation of biotinidase deficiency in infant: High anion gap metabolic acidosis. Karnataka Paediatr J 2021;36(1):57-9.
Abstract
Biotinidase deficiency (BTD) is hereditary autosomal recessive disorder with higher morbidity and mortality if left untreated. We report this case to increase awareness about BTD, presenting with infantile seizures, encephalopathy with high anion gap metabolic acidosis, eczema and to emphasize the importance of early diagnosis in reversal of metabolic acidosis and seizures refractory to multiple anticonvulsants with biotin replacement.
Keywords
Biotinidase deficiency
Multiple carboxylase deficiency
Metabolic acidosis
Infantile seizures

INTRODUCTION
Biotinidase deficiency (BTD) is genetic disorder with autosomal recessive inheritance, in which the body is unable to recycle the vitamin biotin. BTD is of two types: Partial type in which enzyme activity is between 10 and 30% and <10% in severe form.[1,2] BTD is also known as multiple carboxylase deficiency due to deficiency of holocarboxylase synthetase (HCS) (e.g., pyruvate carboxylase) or BTD. The course of BTD can be protracted with intermittent exacerbation of chronic lactic acidosis leading to respiratory distress and other symptoms as seen in this baby. The symptoms may appear as early as 1st week following birth until up to 1 year of age. In this baby, the symptoms were noticed at around 1 month of age. Children with untreated severe BTD present with seizures, hypotonia, respiratory distress, hearing and visual deficit, ataxia, skin rashes, alopecia, and fungal infection (candidiasis). Affected children will also have developmental delay. Eczematous skin rash, alopecia, conjunctivitis, candidiasis, and ataxia are the features more specific to severe BTD. Symptoms may appear as early as 1st week following birth until up to 1 year of age. Delayed diagnosis and treatment may cause irreversible neurological damage, growth retardation, and autistic behaviors. The aim of the therapy is to increase biotin bioavailability by daily supplementation of biotin (5–20 mg) for lifetime.[3,4]
CASE REPORT
2-month-old girl born to non-consanguineously married couple, admitted to pediatric intensive care with multiple episodes of seizures since 3 weeks, breathing difficulty, conjunctivitis, eczematous skin rash in flexural areas, and hair loss [Figures 1 and 2]. Physical examination of an afebrile baby revealed tachycardia, signs of poor perfusion, altered sensorium, conjunctivitis, and deep respiratory efforts with grunting. In view of impending respiratory failure and Glasgow coma scale <8/15, child required invasive ventilation.
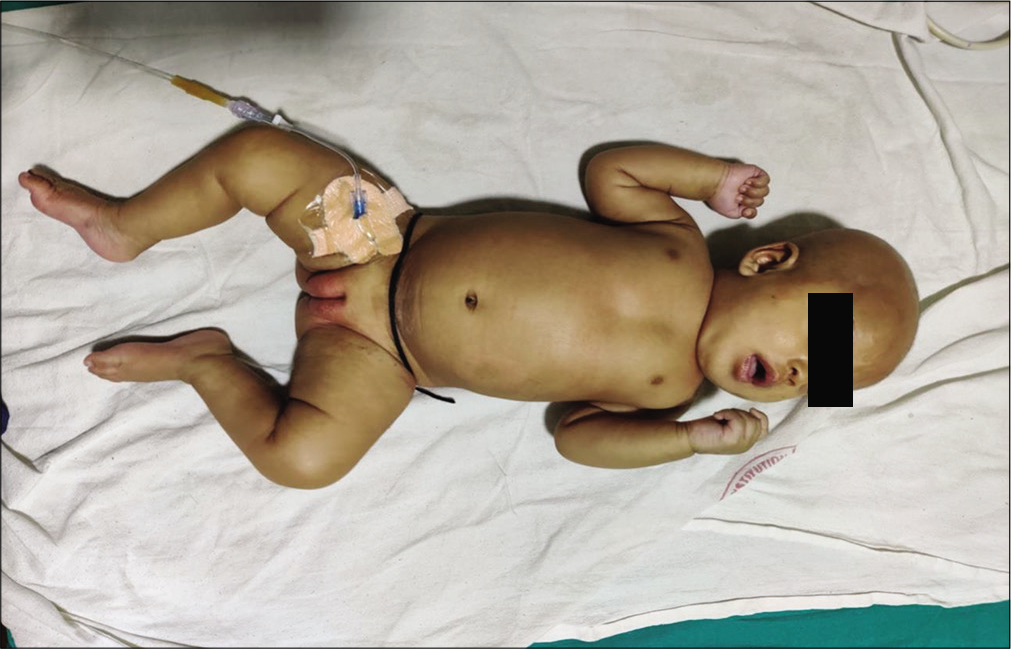
- Two-month-baby girl presented with hypotonia, seizures, alopecia, conjunctivitis, and skin rash.
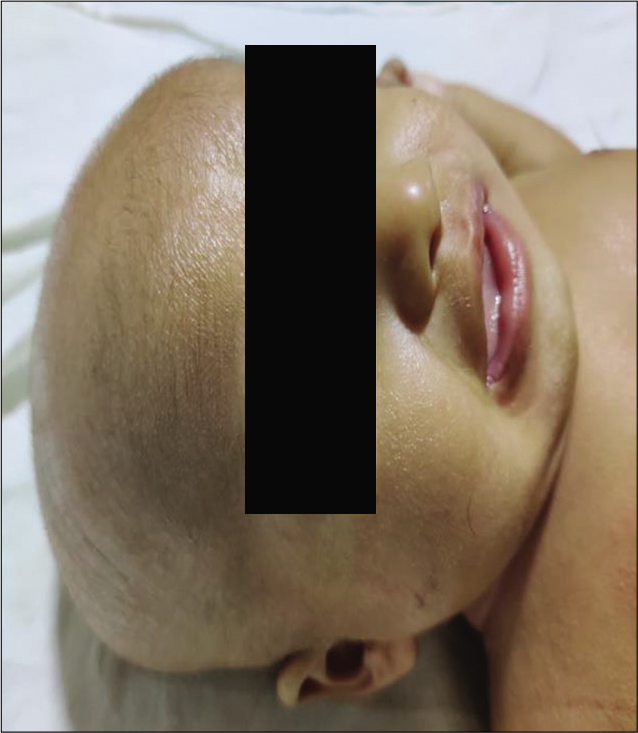
- Baby with alopecia, conjunctivitis, and dry skin.
Investigations done showed normocytic normochromic blood picture with neutrophilic leukocytosis and thrombocytosis. Blood gas showed high anion gap metabolic acidosis, with high arterial lactates and increased levels in serum ammonia. Patient was resuscitated with fluid for shock, treated with bicarbonate correction in view of metabolic acidosis and multiple anticonvulsants. Electroencephalograph showed poorly formed background activity with right hemispheric slowing pattern, neurosonography was normal, in view of suspicion of IEM due to refractory seizures, persistent metabolic acidosis, and IEM specific medications (mitochondrial cocktail) along with antibiotics were started. Ophthalmology evaluation done showed hypermetropic silk shot fundus with refractive error and no optic atrophy. BERA showed bilateral severe to profound hearing loss [Figure 3].
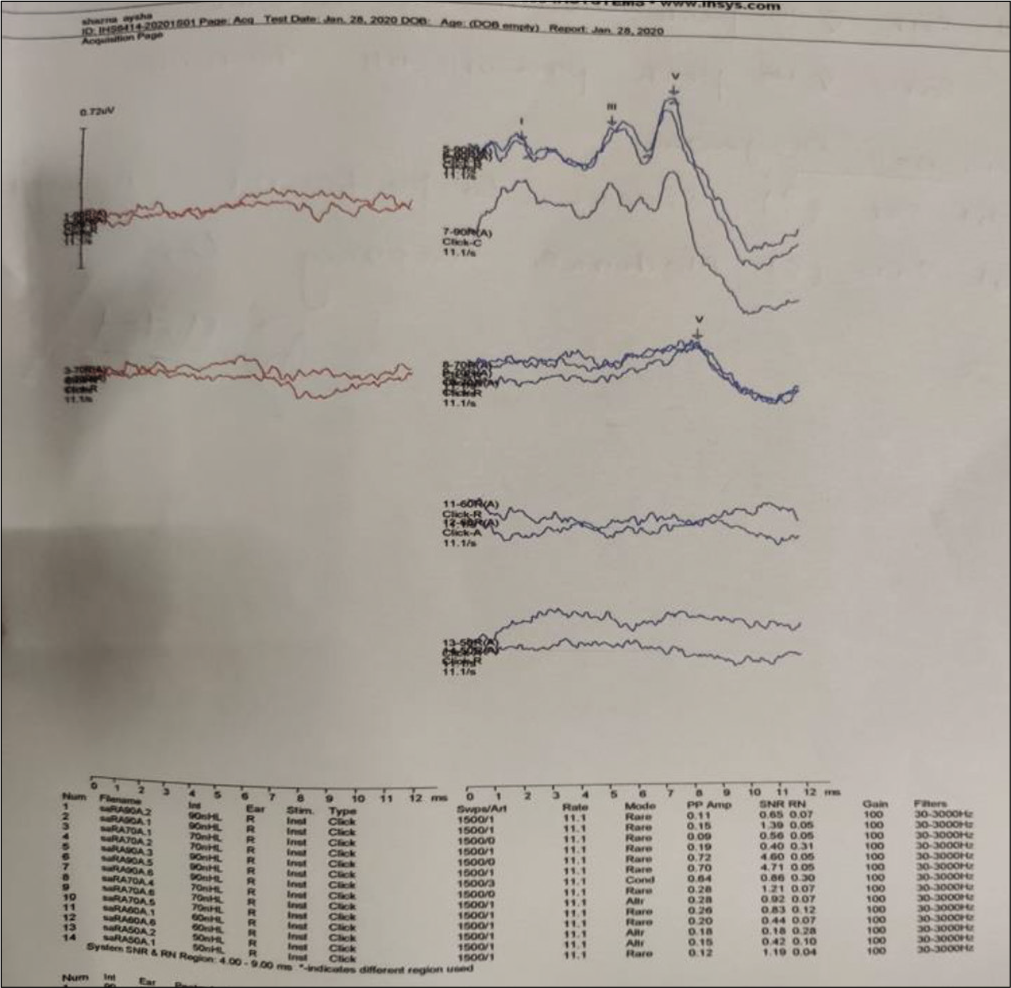
- BERA showing right ear V peak absent till 90 Db, left ear V peak present till 70 Db.
Patient was suspected to have IEM possibly organic academia and BTD. TMS sent in view of suspected IEM showed BTD (enzyme activity of 25.4U against normal value 31.6–388U) and high levels of acylcanitines (methylmalonyl carnitine, and 3-OH-Isovalerayl carnitine deficiency). Urinary organic acids analyzed were within normal limits. Molecular analysis/genetic testing was not performed due to parental disagreement to consent for the test. Child was started on twice daily dose of oral biotin replacement (dose of 10 mg/day) along with anticonvulsants (tapered gradually over time) which showed dramatical improvement in her symptoms, that is, encephalopathy, respiratory distress, and persistent metabolic acidosis. The need for regular neuro-developmental follow-up and the risk of recurrence of symptoms in case of non-compliance to treatment has been explained to parents.
DISCUSSION
Biotinidase is an enzyme which acts by releasing biotin from dietary proteins. BTD is genetic disorder with autosomal recessive inheritance. Mutations of BTD gene cause BTD. The BTD gene is required for production of the enzyme-biotinidase. This enzyme recycles biotin, Vitamin B found in foods (liver, egg yolks, and milk products). Biotinidase converts dietary protein bound biotin to free (active) form. Enzymes like biotin-dependent carboxylases make use of free biotin to metabolise fats, proteins, and carbohydrates.[4] Several enzymes are impaired in BTD, the condition is named as multiple carboxylase deficiency. Impaired biotin availability causes multiple carboxylase deficiency leading to secondary ketoacidosis, hyperammonemia, and organic aciduria. Neurocutaneous symptoms such as seizures, motor mental retardation, spastic paraparesis, ataxia, hypotonia, sensorineural deafness, optic atrophy, eczematous dermatitis, and alopecia are associated with BTD.[5,6] Detection of low enzyme activity and genetic mutation analysis along with clinical symptoms are diagnostic.[5] The enzyme activity is <10% in severe forms whereas between 10% and 30% in partial deficiency. BTD is a mimicker of primary immunodeficiency. Urine organic acid analysis is normal or increased with 3-hydroxyisovalericacid and 3-methylcrotonylglycine. Plasma acylcarnitine analysis may show normal or increased C5-OH acylcarnitine levels. Biotin replacement may improve symptoms except hearing deficits.[6-10]
We report this case to increase awareness about BTD in setting of infantile onset seizures, encepahalopathy with high anion gap metabolic acidosis, eczema and to emphasize the importance of early diagnosis and reversal of metabolic acidosis, seizures with biotin replacement and antibiotics.
CONCLUSION
Every effort should be made in early diagnosis of biotinidase deficiency , as it is treatable and reversible .Prompt treatment with biotin supplements carries good outcome,as in our case.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Valine, leucine, isoleucine, and related organic acidemias In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson Textbook of Pediatrics (20th ed). Philadelphia, PA: WB Saunders; 2015. p. :649-58.
- [Google Scholar]
- Incidence of biotinidase deficiency in Turkish newborns. Acta Paediatr. 1998;87:1102-3.
- [CrossRef] [PubMed] [Google Scholar]
- Inheritable biotin-treatable disorders and associated phenomena. Ann Rev Nutr. 1986;6:317-43.
- [CrossRef] [PubMed] [Google Scholar]
- Biotinidase deficiency: The enzymatic defect in late onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131:273-81.
- [CrossRef] [Google Scholar]
- Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet Med. 2010;12:464-70.
- [CrossRef] [PubMed] [Google Scholar]
- Disorders of biotin metabolism In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 1989. p. :2083-103.
- [Google Scholar]
- Quantitative determination of biocytin in urine of patients with biotinidase deficiency using high performance liquid chromatography (HPLC) Clin Chim Acta. 1988;177:253-70.
- [CrossRef] [Google Scholar]
- Biotinidase deficiency: Initial clinical features and rapid diagnosis. Ann Neurol. 1985;18:614-7.
- [CrossRef] [PubMed] [Google Scholar]
- Biotinidase deficiency: A survey of 10 cases. Arch Dis Child. 1988;63:1244-9.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebral metabolic change after treatment in biotinidase deficiency. J Inherit Metab Dis. 1993;16:399-407.
- [CrossRef] [PubMed] [Google Scholar]




