
Translate this page into:
Cornelia de Lange syndrome: A novel case report
*Corresponding author: Prithviraj Ramakrishna, Department of Pediatrics, M.V. Jayaram Medical College and Research Hospital, Bengaluru, Karnataka, India. prithviraj9686@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Ramakrishna P, Arun M, Vasudevan N. Cornelia de Lange syndrome: A novel case report. Karnataka Paediatr J. doi: 10.25259/KPJ_14_2025
Abstract
Cornelia de Lange syndrome (CdLS) is a congenital disorder marked by distinctive facial features, severe growth restriction, cognitive disability, global developmental delay and anomalies involving multiple body organs. Majority cases of CdLS are caused due to sporadic mutations in the NIPBL, SMC1A, SMC3, RAD21 or HDAC8 genes, which form/regulate a multiprotein complex called cohesin. Cohesin is required for the separation of sister chromatids during cell division. Here, we present a rare case of a 3 years 6 months depicting classical features of CdLS with missense mutation c.932c>T in the HDAC8 gene with mild spasticity in all four limbs and speech delay.
Keywords
Cornelia de Lange syndrome
HDAC8
Missense mutation

INTRODUCTION
Cornelia de Lange syndrome (CdLS) was first described as a distinct syndrome in 1933, by Dr. Cornelia de Lange, a Dutch Paediatrician, after whom the disorder has been named. This is a congenital anomaly syndrome characterised by facial dysmorphism, primordial short stature, hirsutism and upper-limb reduction defects. Craniofacial features include synophrys, arched eyebrows, long eyelashes, widely spaced teeth and microcephaly. Many children demonstrate autistic and self-destructive tendencies. It is an autosomal-dominant disorder caused by specific gene mutations and occurrence is 1 in 30000–50000 children. The precise prevalence of the disease is unknown but is estimated to be 1–10:100,000.[1] Depending on the mutated gene, CdLS can be inherited in an autosomal-dominant manner when it is caused by variations in the NIPBL, SMC2 or RAD21 genes, or it can have an X-linked inheritance when variations in the SMC1A or HDAC8 genes cause it.[2] However, most cases (more than 99%) result from new (de novo) mutations. with certain features of systemic involvement, particularly congenital anomalies of the heart or throat.[2]
CASE REPORT
A 3-year-old 6-month-old female proband who was a 2nd born to a non-consanguineous marriage with none of the family members having similar complaints, was brought to the outpatient department with complaints of multiple flat dark and white lesions over the trunk since birth and recurrent respiratory tract infection. The mother also noticed the child had shown a delay in motor milestones in that she was not able to stand and walk without support, she was not able to speak two word phrases and did not respond to name calls, doing gestures for her needs. The proband was born full term by normal vaginal delivery with birth weight of 2.4 kg, cried immediately at birth after which she developed complications like meconium aspiration which required neonatal intensive care unit (NICU) admission at the first 3 days of life. On 33 days of life, she developed recurrent vomiting and breathing difficulties and was diagnosed with aspiration pneumonia, and ultrasonography (USG) abdomen showed congenital hypertrophic pyloric stenosis for which she underwent laparoscopic pyloromyotomy and conservatively managed. At 3 months of age, 2D ECHO was done which revealed that she had an ostium secundum atrial septal defect.
On clinical examinations, the patient showed dysmorphism with brachy microcephaly (head circumference [HC] = 42 cm, <3rd centile), triangular face with pointed chin, bushy eyebrows, synophrys, depressed nasal bridge, long philtrum, high arched palate, low set of ears, skin hypopigmentation [Figure 1a and b] and short stature (height = 82 cm, <3rd centile), weight (10 kg, <3rd centile), delayed eruption of teeth with widely spaced teeth with growth showed osseous retardation. There was mild spasticity in all four limbs with normal reflexes, along with micromelia of hands and feet. On evaluation, the child had a normal routine blood investigation, tandem mass spectrometry (TMS) and lactate - normal, chest X-ray and magnetic resonance imaging brain were normal with no gross abnormalities, her audiological testing, brainstem evoked response audiometry done at 1 year 1 month suggestive of right and left profound sensorineural hearing loss. Left wrist radiograph for age estimation at 3 years 6 months showed reciprocal shaping of Capitate and Hamate was visualised but the complete absence of ossification centres of the scaphoid, lunate, triquetral, trapezoid with the bone age of >4 months but <2 year suggestive of bone age less than chronological age [Figure 2].

- (a) Hypopigmented patches since birth. (b) Dysmorphic facies with triangular face and low set of ears.
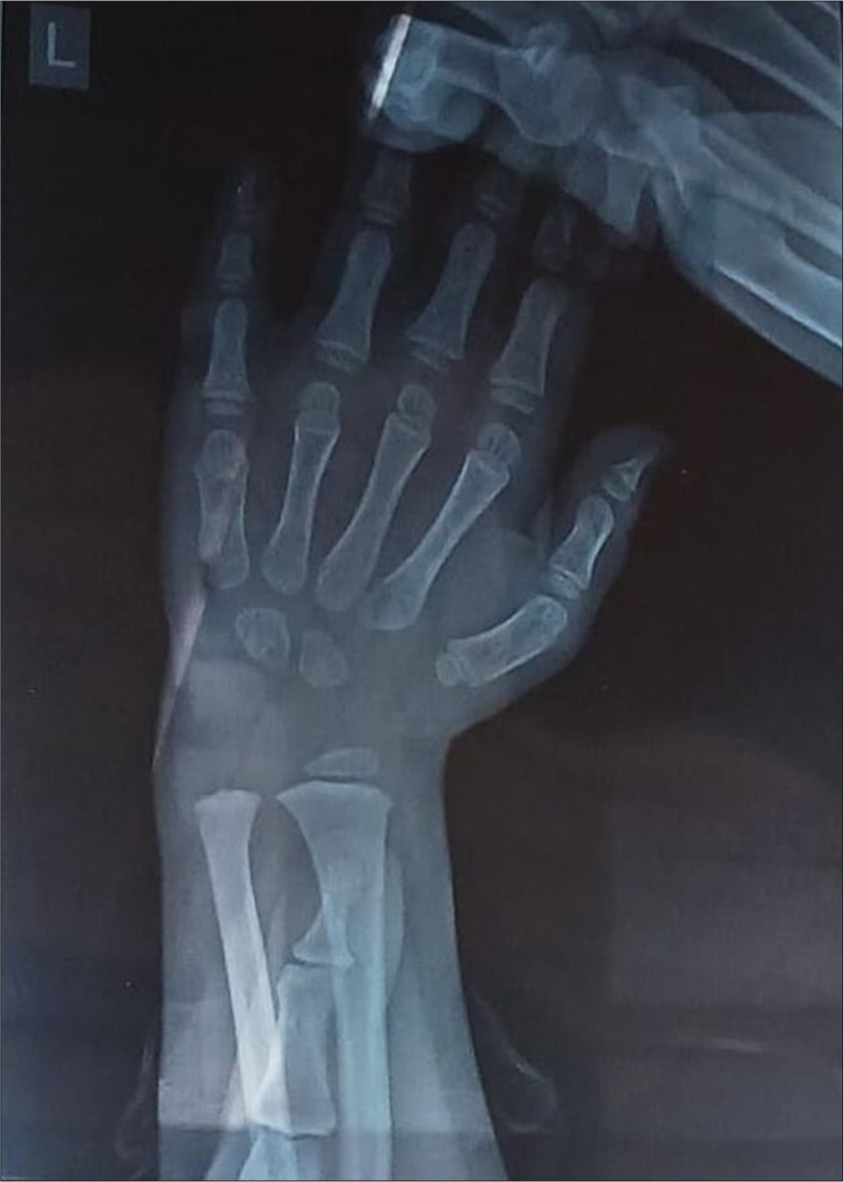
- Left hand wrist X-ray showing only two carpal bones.
On further evaluation, clinical exome sequencing of proband done at 2½ years resulted in HDAC8 variant Missense mutation, exon 9, HDAC8 (ENTST00000373573.9): c.932C>T (p.Thr311Met), heterozygous-linked dominant inheritance pathogenic. On sanger sequencing of proband gene for parents, showed carrier for the same in mother [Table 1]. Other members of the family were not affected by disease causing genes.
| Details | Gene | Location | Variant | Zygosity | Disease | Inheritance | Classification |
|---|---|---|---|---|---|---|---|
| Proband | HDAC8(-) | Exon 9 | c.932C >T (P.Thr311Met) | Heterozygous | Cornelia-de lange syndrome 5 | X-linked dominant | Pathogenic |
| Mother | HDAC8(-) | Exon 9 | c.932C >T (P.Thr311Met) | Heterozygous | Carrier | ||
| Father | HDAC8 | - | - | - | absent | - | - |
DISCUSSION
CdLS is caused by mutation in gene HDAC8 on chromosome 9 with X linked dominant inheritance, they present with delayed anterior fontanelle closure and more pronounced ocular hypertelorism. There are very few cases reported with similar findings. The index phenotype child here is similar to the 4-year-old reported back in the year 2018 showing growth retardation with gastroesophageal reflux.[3] We observed that our child had features of dysmorphisms as reported in CdLS-5 patients, including arched eyebrows, depressed nasal bridge and long philtrum. The index child had no limb anomalies, and skeletal abnormalities a characteristic feature observed in CdLS-5 patients.[4,5]
The phenotype can also be more variable because HDAC8-associated CdLS is X-linked and influenced by random X-inactivation in females. HDAC8-related CdLS is usually caused by point mutations (missense or null) spread throughout the gene. Depending upon the position of these mutations, they determine the residual enzymatic activity, which in turn leads to varying degrees of loss of acetylation and thus contributes to the variable phenotype in patients. The index child’s missense variant c.932c>T is located in the loop region of the HDAC8 protein surrounding the active site causing the loss of the enzymatic function of HDAC8.[2,4]
Management of these children involves a multidisciplinary team of paediatrician, physiotherapist, speech and occupational therapist. The short stature on serial monitoring with delayed puberty can be treated with recombinant human growth hormone. The index child is currently managed by a multidisciplinary team for recurrent respiratory infections and physiotherapy, speech and vocational therapies. The limitation of this report is that the follow-up is short-term in the index child.
CONCLUSION
A phenotypic, multidimensional approach by a team of paediatric neurologist, paediatrician, and dermatologist from various faculties helped to diagnosing the rare condition CdLS helps in the good prognosis of a child into adulthood, serial monitoring of growth and provision of growth hormone therapy will be helpful and has waned significantly but not entirely over time, follow-up is advised.
Ethical approval:
Institutional Review Board approval is not required.
Declaration of patient consent:
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest:
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation:
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Cornelia de-lange syndrome: A case report. Int J Clin Pediatr Dent. 2013;6:115-8.
- [CrossRef] [PubMed] [Google Scholar]
- Cornelia de lange syndrome In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993. [Last accessed on 2020 Oct 15]
- [Google Scholar]
- Cornelia de lange syndrome in a 4-year-old child from India: Phenotype description and role of genetic counseling. Med Arch. 2018;72:297.
- [CrossRef] [PubMed] [Google Scholar]
- A classic cornelia de lange syndrome type 5 (CdLS5) With a de novo missense variation of p.Gly210Arg in the HDAC8 Gene with a novel phenotype of generalized dystonia. Cureus. 2024;16:e60838.
- [CrossRef] [PubMed] [Google Scholar]
- Cornelia de lange syndrome: A rare genetic disorder. Pediatr Rev Int J Pediatr Res. 2020;7:152-6.
- [CrossRef] [Google Scholar]




