
Translate this page into:
Epileptic Spasms-West syndrome secondary to Dravet syndrome due to SCN gene mutation from India
-
Received: ,
Accepted: ,
How to cite this article: Gowda VK, Vegda H, Amoghimath R, Battina M, Shivappa SK, Benakappa N. Epileptic Spasms-West syndrome secondary to dravet syndrome due to SCN gene mutation from India. Karnataka Paediatr J 2021;36(1):49-53.
Abstract
Objectives:
West syndrome (WS) is a triad of epileptic spasms, developmental delay/regression, and hypsarrhythmia. SCN related epileptic encephalopathy is a rare epilepsy syndrome characterized by an early-onset, severe, and epileptic encephalopathy. The causes of WS are multiple and diverse ranging from genetic to structural, metabolic, and unknown causes. The objectives of the study were to report SCN related epileptic encephalopathies with epileptic spasms.
Materials and Methods:
This is retrospective chart review of children presenting with epileptic spasms secondary to SCN gene variants from January 2015 to March 2020 in a tertiary care referral center.
Results:
Out of 15 children, ten were boys. The mean age of presentation was 5 months. Thirteen children had preceded seizures before epileptic spasms in the 1st year of life, two children presented initially with epileptic spasms. No neuro-deficits were noted in all the children. In all the cases electroencephalogram was suggestive of hypsarrhythmia. Routine testing, neuroimaging, and metabolic tests were normal in all the cases. Various pathogenic variants seen in next-generation sequencing were SCN1A in 11, SCN1B and SCN2A in two children each. Three children responded for vigabatrin and five children responded for steroids but all of them had relapse and were refractory to other antiepileptic drugs.
Conclusion:
SCN related epileptic encephalopathy should be considered in the differential diagnosis of epileptic spasms. These infants present earlier compare to classical Dravet syndrome children.
Keywords
Epileptic spasms
West syndrome
SCN mutation
Epileptic encephalopathy

INTRODUCTION
Dravet syndrome (DS) and SCN related encephalopathies are genetically determined severe early onset epileptic encephalopathy (EOEE), which begins in the 1st year of life in an otherwise normal infant.[1] Initial seizures, often induced by a fever, tend to be prolonged generalized or unilateral tonic and/or clonic seizures, are more frequent and come in clusters. Beyond the 1st year, multiple seizure types often develop such as myoclonic, atypical absence, and focal seizures. West syndrome (WS) is the most common epilepsy syndrome in infancy, which is characterized by triad of infantile spasms, developmental deterioration, and hypsarrhythmia.[2] It can be due to structural, genetic, infectious, immune, metabolic and unknown cases. Several genes, such as, SCN1A, SCN2A, SCN1B, ATXN2, NR3C1, KPNA7, STXBP1, ABCB1, GRIN1, ARX, and TSC2, were found to be associated with the pathogenesis of epileptic spasms.[3] Variants in the gene encoding voltage-gated sodium channel (SCN1A) are associated with several epilepsy syndromes. Here, we report children of SCN related epileptic encephalopathies who presented with epileptic spasms.
MATERIALS AND METHODS
This is a retrospective chart review of epileptic spasms due to SCN related epileptic encephalopathies from tertiary care referral center, from southern part of India. The medical records of children attending the pediatric neurology clinic and those who were admitted in pediatric neurology and pediatric ward from January 2015 to March 2020 were analyzed. Among them, only children who were confirmed to have a diagnosis of SCN related epileptic encephalopathies in genetic studies and presented with epileptic spasms were included and formed the study group. Those children with epileptic spasms and suspected SCN related epileptic encephalopathies without genetic confirmation were excluded from the study. The data were extracted as per-designed pro forma. Details of history including birth history, developmental history, clinical features including seizure semiology, precipitation of seizures with fever, investigations such as complete hemogram, liver function, renal function, serum calcium, serum ammonia, serum lactate, arterial blood gas, and neuroimaging MRI of brain were taken. Special investigations such as tandem mass spectrometry (TMS), electroencephalogram (EEG), and genetic analysis were also taken. Statistical analysis was performed with SPSS version 21. The results were analyzed. Ethical clearance was obtained, from institutional ethical committee.
RESULTS
A total of 50 children with SCN related epileptic encephalopathies were seen during this period, out of these 15 (35%) had epileptic spasms. The various clinical features, laboratory findings, and outcome of all the 15 children are mentioned in [Table 1]. Of the 15 children, ten (67%) were boys. All of them presented in 1st year of life except once child, with mean age of presentation being 5 months of age compare to 10 months in children with SCN related epileptic encephalopathies without epileptic spasms. All of them presented with other seizures in the form of focal or generalized seizures along with epileptic spams, and except two children who had only epileptic spasms as per history. Seven children presented with fever triggered seizures and spasms, four children presented with seizures after vaccination and four children had afebrile seizures before onset of spasms. Birth history was normal in all the children. Initial development was normal followed by severe developmental delay in all the children. Neuro imaging and TMS were normal in all the children. EEG showed hypsarrhythmia in all the children. EEG showing modified hypsarrhythmia in [Figure 1]. Targeted next generation sequencing showed SCN1A gene mutation in 11 children, SCN1B and SCN2A in two children each. Spasms were controlled initially with vigabatrin and steroids, but later seizures were refractory to treatment.
| Case number | Sex | Age of onset (m.) | Fever triggered seizures | Seizure type | Last follow-up (mo.) and Dev | Gene | Variant | AED tried | Response to AED |
|---|---|---|---|---|---|---|---|---|---|
| 1. | M | 3 | No | GTCS, ES | 45 NA | SCN1A | Ex16.c.3199G>A/p.Ala1067Thr | VPA, LEV,CZM, TPM, ZSM, VB, ACTH | VB |
| 2. | M | 3 | Yes | Focal, ES | 42 NA | SCN1A | Ex8.c.3199G>A/p.Ala1067Thr | VPA, LEV,CZM, TPM, ZSM, VB, ACTH | ACTH |
| 3. | M | 16 | Yes | ES | 37 Amb Autistic | SCN2A | Ex7.c.823C>T/p.Arg275Ter | VPA, CBZ, LEV, CZM, TPM, ZSM, VB,ACTH | VB |
| 4. | M | 3 | No | GTCS, F, ES | 60 NA | SCN1A | c.6013C>T/p.Arg2005Cys | VPA, SP, LEV, CZM, TPM, ZSM, VB, ACTH | None |
| 5. | F | 2 | Yes | GTCS, F, ES | 13 NA | SCN1A | Ex8.c.3199G>A/p.Ala1067Thr | VPA, LEV, CZM, TPM, ZSM, VB, ACTH | ACTH |
| 6. | F | 3 | Yes | GTCS, F, ES | 07 Amb Autistic | SCN1B | c.253C>T/p.Arg85 Cys | VPA, SP, LEV, CZM, TPM, ZSM, VB, ACTH | SP |
| 7. | F | 3 | Yes | GTCS, F, ES | 48 NA | SCN1A | c.3199G>A/p.Ala1067Thr | VPA, SP, LEV, CZM, TPM, ZSM, VB,ACTH | None |
| 8. | M | 3 | Yes | GTCS, F, ES | 12 Autistic | SCN1A | c.3199G>A/p.Ala1067Thr | VPA, LEV, CZM, TPM, ZSM, VB, ACTH | ACTH |
| 9. | M | 9 | Yes | GTCS, F, ES | 33 NA | SCN2A | c.823C>T/p.Arg275Ter | VPA, CBZ, LEV, CZM, TPM, ZSM, VB, ACTH | CBZ |
| 10. | M | 3 | Yes | GTCS, F, ES | 28 Amb Autistic | SCN1A | c.695G>T/p. Gly232Val | VPA, LEV, CZM, TPM, ZSM, VB, ACTH | ACTH |
| 11. | M | 5 | Yes | GTCS, F, ES | 25 NA Autistic | SCN1A | c.3199G>A/p.Ala1067Thr | VPA, SP, LEV, CZM, TPM, ZSM, VB, ACTH | SP |
| 12. | F | 1 | Yes | GTCS, F, ES | 12 NA Autistic | SCN1A | Ex26.c.4907G>A/p.Arg1636Gln | VPA, SP, LEV,CZM, TPM,ZSM, VB,ACTH | None |
| 13. | F | 7 | Yes | GTCS, F, ES | 21, Amb Autistic | SCN1A | Ex26.c.4855A>G/p.Met1619Val | VPA, LEV, CZM, TPM, ZSM, VB, ACTH | ACTH |
| 14. | M | 6 | Yes | GTCS, F, ES | 24 NA Autistic | SCN1A | Ex15.c.2712dupT/p.Ala905Cysfs | VPA, SP, LEV, CZM, TPM, ZSM, VB, ACTH | None |
| 15. | M | 8 | Yes | ES | 18 NA, Autistic | SCN1B | c.560delG/p.Arg187profs | VPA, LEV, CZM, TPM, ZSM, VB, ACTH | VB |
ACTH: Adrenocorticotropic hormone, AEDs: Antiepileptic drugs, Amb: Ambulatory, CLB: Clobazam, CBZ: Carbamazepine, DEV: Development, ES: Epileptic Spasms, F: Focal, GTCS: Generalized tonic-clonic seizures, LEV: Levetiracetam, NA: Non ambulatory, SP: Stiripentol, VPA: Valproate, TPM; Topiramate, VB: Vigabatrin, ZNS: Zonisamide
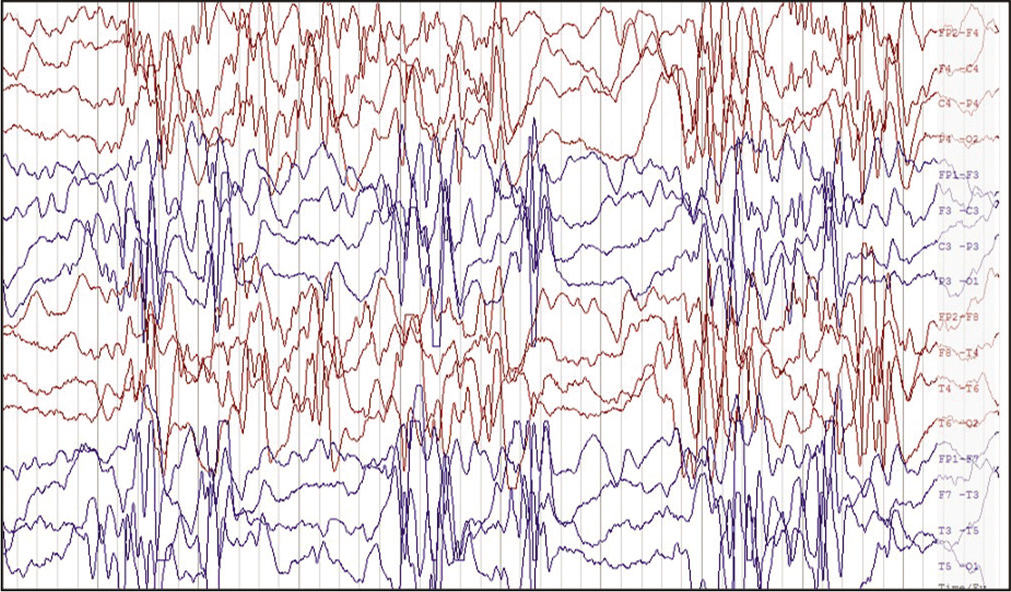
- Electroencephalogram of bipolar longitudinal montage with a sensitivity of 20 μV showing high amplitude multifocal spikes, sharp waves with secondary generalization followed by suppression suggestive of modified hypsarrhythmia in a 8-months-old child with epileptic spasms due to SCN1A pathogenic variant.
DISCUSSION
There are only few reports describing SCN related epileptic encephalopathies presenting with epileptic spasms secondary to mutation in SCN gene in the literature. Here, we are reporting 15 patients presenting as WS with mutation in SCN gene. Around 70–80% of children with SCN related epileptic encephalopathies have point mutations or gross rearrangements in the SCN1A gene. Over 1200 variants associated with epilepsy have been reported in SCN1A.[4] Truncating variants are associated with severe phenotypes. The missense variants are associated with a wide spectrum of phenotypes from DS to much milder forms of epilepsy, such as febrile seizures and “febrile seizures plus,” which are often familial and part of a genetic epilepsy with febrile seizures plus.[5] Ten percent of SCN related epileptic encephalopathy patients have deletion, duplication, or amplification identified with multiplex ligation-dependent probe amplification. In 10–20% of cases, the genetic cause remains unknown, may be other genes are likely to be involved. Less than 1% of SCN related epileptic encephalopathy patients have homozygous mutation in SCN1B, and very few have GABRG2 or SCN2A mutations.[6]
WS is an epileptic encephalopathy with onset typically around 6 months of age, characterized by epileptic spasms, hypsarrhythmia, and developmental delay or regression.
Etiology of epileptic spasms is widely heterogeneous, with acquired and congenital causes. Advances in the genetic investigations have led to discovery of new genes in WS in the past few decades. More than ten genes had been shown to be associated with epileptic spasms.[7] More recently, copy-number variations and mutations in STXBP1, SCN2A, and KCNQ2, which were previously associated with EOEE, have been found in patients with epileptic spasms.[8] Despite these advances, in many cases the cause still remained hidden.[9] The study done by Wallace et al. extends the phenotypic heterogeneity of mutations in SCN1A to include epileptic spasms.[10]
In seven children initial seizures were noticed following DPT vaccine and subsequently developed epileptic spasms, this finding was consistent with other studies of seizures following vaccinations which were reported in 7–57% of children with DS.[11,12] Two children presented with epileptic spasms one had SCN2A and the other with SCN1B mutation.
Ogiwara et al.[13] reported mutation in SCN2A-E1211K, causing epileptic spasms. Nakamura et al.[14] reported, nine of 67 Ohtahara syndrome (OS) cases (13.4%) and one of 150 WS cases (0.67%) were secondary to SCN2A mutation. All nine mutations in patients with OS were in linger regions between two transmembrane segments. In seven of the nine patients with OS, EEG findings transitioned from suppression-burst pattern to hypsarrhythmia.
Harkin et al.[15] described missense SCN1A variant, Nav1.1-p. Thr226Met (T226M) is associated with a far more profound clinical phenotype than typical DS, represents a new class of early infantile epileptic encephalopathy (EIEE) located even beyond DS on the classical severity spectrum of SCN1A-linked disorder. Sadleir et al.[16] described the clinical presentation of more severe SCN1A-linked “early infantile SCN1A encephalopathy.” They identified eight unrelated cases with an identical, presumed de novo missense variants resulting from c.677C > T in SCN1A exon 5. A ninth unrelated child, with the de novo SCN1A missense variant p.Pro1345Ser (c.4033C > T), was also included in the series due to the similarities in symptomology to the T226M patients indicates that early infantile SCN1A encephalopathy can arise from more than one particular variant. In our study, none of them had T226M variant, but most common variant is A1067T.
The mean age of presentation is our study was 5 months; however, mean age of presentation for A1057T variant is 3 months. Mean age of presentation of 50 children with SCN related epileptic encephalopathies with or without epileptic spasms was 10 months. Males are most affected. Only four of them are ambulatory, ten of them had autistic features. Epilepsy is refractory in all children, however epileptic spasms responded for steroids in five and vigabatrin in three children but later relapsed and refractory requiring polytherapy. Six children received stiripentol but only partial response that is more than 50% reduction in seizures noted in two children. We tried carbamazepine, sodium channel blocker in SCN2A subtype but only partial improvement was noted.
Traditional DS can be differentiated from early infantile SCN1A encephalopathy in several ways.[16] Early infantile SCN1A encephalopathy has an earlier age of onset, with seizures arising at an average of 9 weeks of age, more profound developmental impairments; and the majority required feeding tubes. Early infantile SCN1A encephalopathy presents with hyperkinetic movements, as early as 9 weeks of age and epileptic spasms, neither of which are seen in patients with DS, while hyperkinetic movements are not characteristic of SCN1A linked DS. However, similar movements are described with SCN2A and SCN8A-linked EIEEs; this overlap in symptomology led Sadleir et al. to speculate that the early infantile SCN1A encephalopathy, such as SCN2A and SCN8A-linked EIEEs, may be associated with a gain-of-function variant.
Epileptic spasms can be present in SCN mutations. They present earlier than classical DS with more severe developmental delay and refractory to treatment. Most common pathogenic variant noted in this study is A1057T in SCN1A.
CONCLUSION
In all cases of unexplained epileptic spasms, one should consider possibility of SCN gene mutation and genetic testing should be considered. Most common type of SCN1A mutation is A1057T variant. Early identifications are useful to select antiepileptic drugs for this subgroup of epileptic spasms.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Early-onset epileptic encephalopathies and the diagnostic approach to underlying causes. Korean J Pediatr. 2015;58:407-14.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile spasms: A US consensus report. Epilepsia. 2010;51:2175-89.
- [CrossRef] [PubMed] [Google Scholar]
- SCN1A testing for epilepsy: Application in clinical practice. Epilepsia. 2013;54:946-52.
- [CrossRef] [PubMed] [Google Scholar]
- From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 2000;26:13-25.
- [CrossRef] [Google Scholar]
- The SCN1A mutation database: Updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat. 2015;36:573-80.
- [CrossRef] [PubMed] [Google Scholar]
- Sodium channel mutations and epilepsy In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, DelgadoEscueta AV, eds. Jasper's Basic Mechanisms of the Epilepsies. Bethesda, MD: National Center for Biotechnology Information; 2012. p. :675-87.
- [Google Scholar]
- Infantile spasms syndrome, west syndrome and related phenotypes: What we know in 2013. Brain Dev. 2014;36:739-51.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile spasms are associated with abnormal copy number variations. J Child Neurol. 2013;28:1191-6.
- [CrossRef] [PubMed] [Google Scholar]
- Whole-exome sequencing improves the diagnosis yield in sporadic infantile spasm syndrome. Clin Genet. 2016;89:198-204.
- [CrossRef] [PubMed] [Google Scholar]
- Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. 2003;61:765-9.
- [CrossRef] [PubMed] [Google Scholar]
- A retrospective study of the relation between vaccination and occurrence of seizures in dravet syndrome. Epilepsia. 2011;52:175-8.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence of SCN1A-related dravet syndrome among children reported with seizures following vaccination: A population-based ten-year cohort study. PLoS One. 2013;8:e65758.
- [CrossRef] [PubMed] [Google Scholar]
- De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73:1046-53.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical spectrum of SCN2A mutations expanding to ohtahara syndrome. Neurology. 2013;11:992-8.
- [CrossRef] [PubMed] [Google Scholar]
- The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130:843-52.
- [CrossRef] [PubMed] [Google Scholar]
- Not all SCN1A epileptic encephalopathies are dravet syndrome: Early profound Thr226Met phenotype. Neurology. 2017;89:1035-42.
- [CrossRef] [PubMed] [Google Scholar]




