
Translate this page into:
Expanding the phenotypic spectrum of Rauch–Steindl syndrome: A case study of a novel NSD2 mutation in an Indian patient
*Corresponding author: Mohd Faisal Zoheb, Department of Paediatrics, Mahadevappa Rampure Medical College, Kalaburagi, Karnataka, India. faisalzoheb007@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Zoheb MF, Patil A, Kinagi AB, Nemadi ND. Expanding the phenotypic spectrum of Rauch–Steindl syndrome: A case study of a novel NSD2 mutation in an Indian patient. Karnataka Paediatr J. 2024;39:110-4. doi: 10.25259/KPJ_25_2024
Abstract
Rauch–Steindl syndrome (RAUST) is a rare genetic disorder characterised by poor growth, distinctive facial dysmorphisms and variable developmental delays. It is a milder variant of Wolf–Hirschhorn syndrome and is associated with mutations in the NSD2 gene. This study presents a case of RAUST with a novel NSD2 mutation, expanding the phenotypic spectrum of the syndrome. An 18-month-old Indian boy was referred to Basaveshwar Teaching and General Hospital for failure to thrive. Born preterm at 1.7 kg with intrauterine growth restriction, he presented with severe growth retardation, microcephaly and facial dysmorphism including dolichocephaly, triangular facies and cleft palate. Developmental assessment revealed significant delays in motor and speech milestones. Genetic analysis identified a heterozygous NSD2 gene variant (chr 4: g.1978839del; p.Pro1343GlnfsTer49), leading to a frameshift and premature truncation of the protein. Whole exome sequencing was performed on peripheral blood DNA, revealing the pathogenic NSD2 mutation. The analysis also highlighted the variability in phenotypic expression of RAUST, including additional anomalies such as undescended testes. The proband’s clinical features align with RAUST, with the identified NSD2 mutation contributing to the observed dysmorphisms and developmental delays. Despite normal neuroimaging, the wide spectrum of symptoms, including additional anomalies, underscores the complex phenotypic variability of RAUST. The mother’s similar phenotypic features suggest possible autosomal dominant inheritance with variable expressivity. This case highlights the diverse presentation of RAUST and the critical role of NSD2 mutations in its pathogenesis. It emphasises the need for genetic analysis in diagnosing RAUST and understanding its broad phenotypic spectrum. Further studies are necessary to explore the full range of clinical manifestations and inheritance patterns associated with RAUST.
Keywords
Rauch–Steindl syndrome
NSD2 gene mutation
Developmental delay
Dysmorphic features
Whole exome sequencing

INTRODUCTION
Rauch–Steindl syndrome (RAUST) is characterised by poor pre- and postnatal growth, sometimes with short stature and small head circumference, characteristic dysmorphic facial features and variable developmental delay with delayed motor and speech acquisition and impaired intellectual function that can be mild.[1] Other features may include hypotonia and behavioural abnormalities.[1] The phenotype represents a mild form of Wolf–Hirschhorn syndrome (WHS; 194190), which is a contiguous gene deletion syndrome caused by heterozygous deletion of several genes on chromosome 4p16.[2] The clinical features of RAUST are similar to but milder than those of WHS, with less severe dysmorphic facial features, less severe developmental disabilities in general and absence of a seizure disorder.[2] The phenotype and expressivity of RAUST are highly variable.[3]
The heterozygous mutations in the NSD2 gene that were identified in patients with RAUST by Lozier et al. (2018), Derar et al. (2019) and Barrie et al. (2019) occurred de novo.[3-5] The transmission pattern of RAUST in the Chinese family reported by Hu et al. (2020) was consistent with autosomal dominant inheritance with incomplete penetrance and variable expressivity.[6] Most of the heterozygous mutations in the NSD2 gene identified in RAUST patients by Zanoni et al. (2021) occurred de novo.[7] However, there were 2 families (families 6 and 7) in which the transmission pattern was consistent with autosomal dominant inheritance.[7]
Patients with RAUST exhibit a wide range of mild phenotypic features, with core manifestations of microcephaly, intrauterine growth restriction, facial dysmorphisms, autism, intellectual disability, low birth weight, feeding difficulties, failure to thrive, short stature, speech delay and muscular hypotonia.[8] In this study, we identified a heterozygous NSD2 gene variant (chr 4: g.1978839del; Depth:86x) that results in a frameshift and premature truncation of the protein 49 amino acids downstream to codon 1343 (p.Pro1343GlnfsTer49; ENST00000508803.6) [Figure 1] in an Indian boy diagnosed with RAUST. In addition, our study further expands the phenotypic spectrum of RAUST with genetic mutations and associated maternal phenotype.[9]
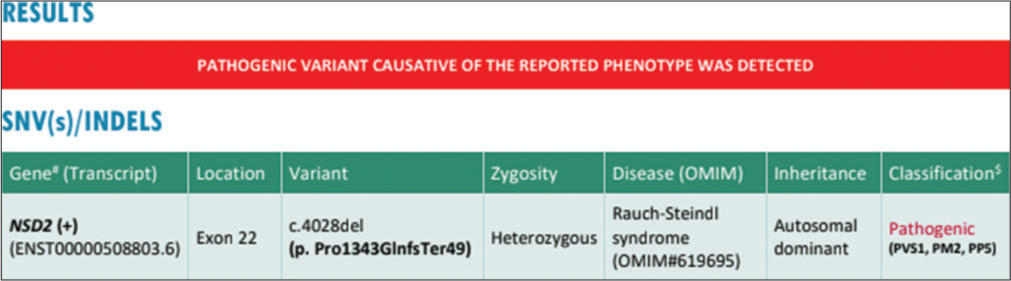
- Genetic report, Whole Exome Sequencing showing pathogenic variant of Rauch-Steindl syndrome (red color), SNV: Single Nucleotide Variant, INDEL: Insertion or Deletion, OMIM: Online Mendelian Inheritance in Man, NSD2: NSD2 nuclear receptor binding SET domain protein 2, PVS1: Pathogenic very strong, PM2: Pathogenic moderate, PPS: Pathogenic supporting. #: The in-silico prediction# of the variant, $: Due to inadequate literature evidence, this NSD2 variation is classified as a pathogenic variant and has to be carefully correlated with the clinical symptoms.
CASE REPORT
Patient
An 18-month-old was referred to Basaveshwar Teaching and General Hospital Kalaburagi for failure to thrive on 29th April 2024. The proband’s parents provided written informed consent for the publication of photographs as well as clinical and genetic data. The present study was approved by Department of Paediatrics, Basaveshwar Teaching and General Hospital attached to M R Medical College, Kalaburagi.
Genetic analysis
Whole exome analysis
Genomic DNA was extracted from 2-mL peripheral blood samples from the proband. Whole exome sequencing was performed at MedGenome Labs Ltd., Bangalore. DNA extracted from blood was used to perform targeted gene capture using a custom capture kit. The libraries were sequenced to mean depth of >80-100X on Illumina sequencing platform. The sequences obtained are aligned to human reference genome (GRCh38) using BWA aligner [Sentieon] and analysed using Sentieon for removing duplicates, recalibration and re-alignment of indels [Sentieon]. Sentieon haplotype caller is then used to identify variants in the sample. The germline variants identified in the sample is deeply annotated using VariMAT pipeline. In addition to single nucleotide variant (SNV) and small Indels, copy number variants are detected from targeted sequence data using the ExomeDepth method.
RESULTS
Clinical description
The proband [Figure 2] is an 18-month-old boy born to nonconsanguineous parents, admitted for delayed development and growth. He was born late preterm with intrauterine growth restriction (birth weight: 1.7 kg), accompanied by cleft palate and respiratory distress, requiring a 6-day neonatal intensive care unit stay. Initial concerns included low birth weight and feeding difficulties due to the cleft palate.
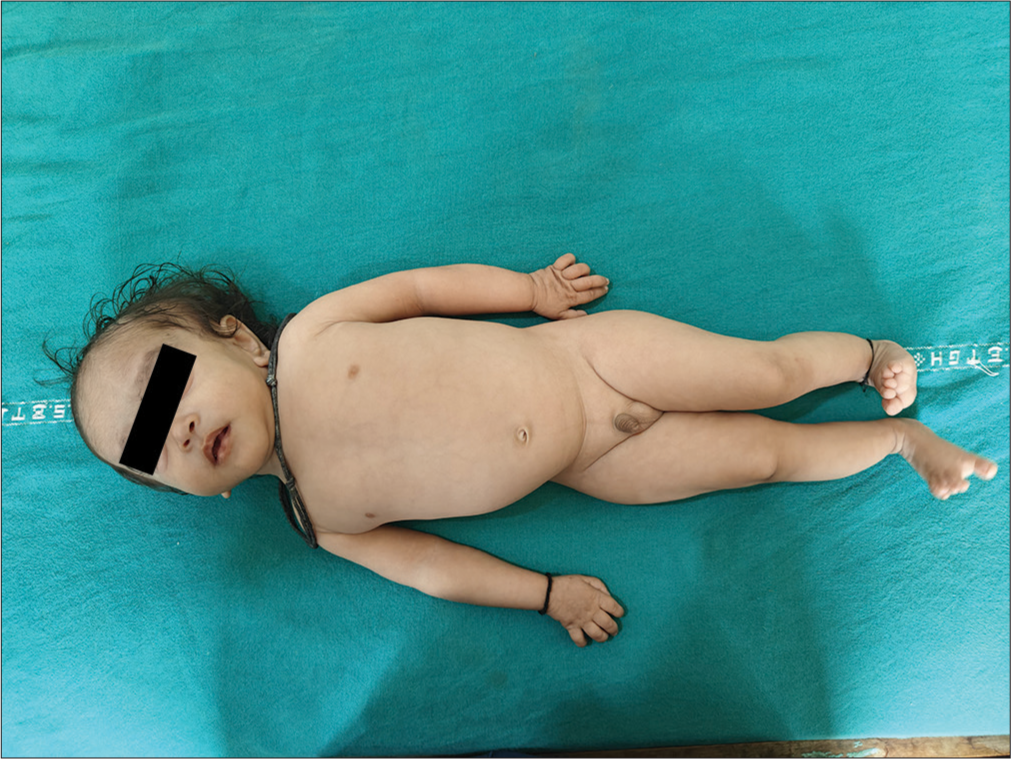
- Proband.
Developmentally, at 7 months, he had a partial neck hold, social smile and alertness to sounds. By 8 months, he could roll over and reach for toys. At 17 months, he sits with support, has an immature pincer grasp, coos and recognises his mother. His developmental quotient (DQ) is 13.8% for his age.
At present, he has severe acute malnutrition (weight: 2.7 kg, <−3 standard deviation [SD]) and severe short stature (length: 52 cm, <−3 SD). Table 1 depicts comparison of proband features with a reference case by Yang et al.[10] Dysmorphic features include microcephaly (head circumference: 35 cm, <−3 SD), dolichocephaly, wide-open anterior fontanelle, broad forehead, triangular facies, eccentric pupils, prominent ears, deep-set eyes, periorbital fullness, flat nasal bridge, micrognathia, delayed dentition, widely spaced nipples, delayed bone age and undescended testes.
| Clinical Features | Patient | Reference Case (Yang et al) |
|---|---|---|
| Variant | p.Pro1343GlnfsTer49 | c.2721delT(p.Asn907Lysfs*5) |
| Gender | Male | Female |
| Age at examination | 18 months | 7 years |
| Gestation | 36 weeks (Late preterm) | Full-term |
| Birth weight | 1.7 kg | 2.15 kg (<−2.5 SD) |
| Birth length | Not provided | 45 cm (<−2 SD) |
| Feeding difficulty | Yes | Yes |
| Hypotonia | Yes | Yes |
| Weight | 2.7 kg (<-3 SD) | 15 kg (<−3 SD) |
| Length/Height | 52 cm (<−3 SD) | 118 cm (<−1 SD) |
| Head circumference (OFC) | 35 cm (<−3 SD) | 49 cm (<−2 SD) |
| Developmental delay | Yes, global developmental delay | Yes, cognitive impairment, delayed motor milestones |
| Age of walking | Not reached (18 months) | 20 months |
| Age of first words | Not reached | 25 months |
| Brain anomalies | MRI brain – Normal | MRI normal |
| Facial dysmorphism | Microcephaly, dolichocephaly, wide-open anterior fontanelle, broad forehead, triangular facies, eccentric pupils, prominent ears, deep-set eyes, periorbital fullness, wide/flat nasal bridge, cleft palate, micrognathia, delayed dentition | Triangular face, proptosis, hypertelorism, broad forehead, high anterior hairline, short/upslanted palpebral fissures, sparse eyebrows, short philtrum, micrognathia, abnormal teething |
| Other anomalies | Widely spaced nipples, bilateral undescended testes | Small hands/feet, mild clinodactyly of the right hand, loose skin on hands/feet |
MRI: Magnetic resonance imaging, SD: Standard deviation, OFC: Occipito-Frontal Circumference
Investigations (magnetic resonance imaging [MRI] brain, spine X-ray, chest X-ray [Figure 3], echocardiography, abdominal ultrasonography and brainstem evoked response audiometry (BERA)) were normal.
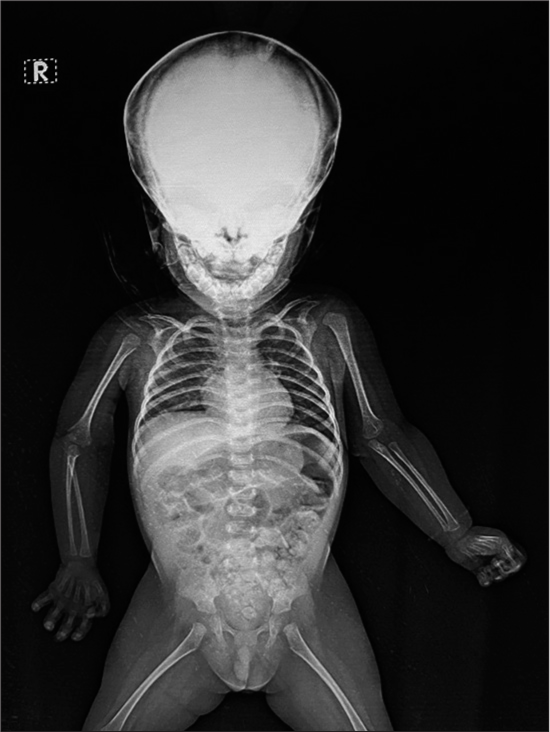
- Skeletal survey.
The mother [Figure 4] exhibits phenotypic traits of RAUST, including low birth weight, short stature, triangular facies, wide forehead, micrognathia, strabismus and refractive errors.
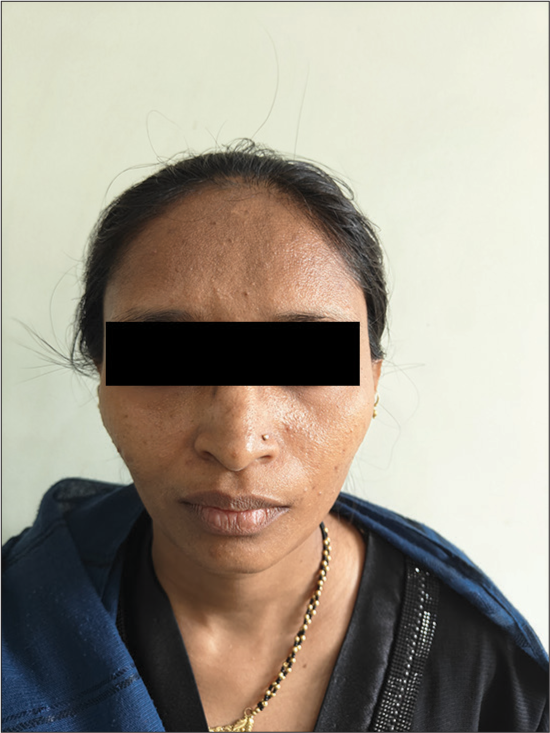
- Proband’s mother.
DISCUSSION
Clinical presentation and dysmorphic features
The proband exhibits multiple dysmorphic features such as microcephaly, dolichocephaly, triangular facies, deep-set eyes and cleft palate. These are consistent with RauchSteindl syndrome (RSS), as described in the literature. The presence of less commonly discussed features, such as bilateral eccentric pupils and prominent ears might reflect the phenotypic variability within RSS or an additional genetic anomaly. The NSD2 variant identified in the proband has been linked to developmental phenotypes similar to those observed in RSS, suggesting that the genetic mutation directly contributes to craniofacial and developmental abnormalities.[1]
Growth parameters and development
The child’s severe growth retardation and global developmental delay align with the typical RSS presentation. The proband’s DQ of 13.8%, severe hypotonia and delayed motor milestones (e.g., sitting with support at 17 months) are characteristic of the developmental challenges associated with pathogenic NSD2 mutations. The identified NSD2 variant, known for causing frameshifts and truncation, likely disrupts normal protein function, leading to the observed growth and developmental delays. The literature supports that truncating mutations in NSD2 are a significant cause of the profound growth and developmental impairments seen in RSS.[2]
Genetic findings
The heterozygous 1 base pair deletion in the NSD2 gene (p.Pro1343GlnfsTer49) is a critical finding that correlates with the proband’s clinical presentation. This specific mutation has been reported in similar cases, further supporting its pathogenic role. NSD2 mutations are well-documented in RSS cases, particularly those involving frameshifts and premature truncations, which lead to the loss of essential protein functions. The proband’s mutation is classified as pathogenic, consistent with other reported cases where NSD2 mutations result in the developmental and dysmorphic features characteristic of RSS.[11]
Brain and organ anomalies
Despite the significant developmental delays, the proband’s brain MRI and other organ evaluations are normal. This is consistent with many RSS cases where neuroimaging does not reveal structural anomalies despite severe developmental issues. The identified NSD2 mutation, while leading to profound developmental delays, may not necessarily cause structural brain anomalies, as seen in the proband. The literature suggests that functional disruptions due to NSD2 mutations primarily manifest as neurodevelopmental delays rather than structural abnormalities.[12]
Additional anomalies
The proband exhibits bilateral undescended testes and widely spaced nipples, features that are less commonly associated with RSS but may represent an extended phenotypic spectrum due to the NSD2 mutation. The variability in phenotypic expression, including these additional anomalies, might be due to the specific truncating mutation in the NSD2 gene, highlighting the broad spectrum of clinical manifestations that can arise from such mutations.[13]
CONCLUSION
This case of an 18-month-old boy with a heterozygous NSD2 gene variant (p.Pro1343GlnfsTer49) provides valuable insights into RAUST. The patient’s clinical presentation, including microcephaly, developmental delay and dysmorphic features, aligns with RAUST. The identified NSD2 mutation correlates with severe growth and developmental challenges, consistent with previously reported cases. Notably, the proband’s mother also exhibits phenotypic features associated with RAUST, such as low birth weight, short stature and facial dysmorphisms, suggesting a possible autosomal dominant inheritance pattern with variable expressivity. Despite normal neuroimaging and additional anomalies like undescended testes, the findings highlight the broad phenotypic spectrum of RAUST and reinforce the role of NSD2 mutations in its pathogenesis. This case underscores the importance of genetic analysis in diagnosing RAUST and understanding its clinical variability.
Ethical approval
The Institutional review board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Clinical features of Rauch-Steindl syndrome: Overview and case reports. Am J Med Genet. 2001;98:51-60.
- [Google Scholar]
- Genetic analysis of patients with Rauch-Steindl syndrome (RAUST): Expanding the phenotypic spectrum. Eur J Hum Genet. 2021;29:231-40.
- [Google Scholar]
- A case study of Rauch-Steindl syndrome (RAUST) with emphasis on de novo NSD2 mutations. J Med Genet. 2019;56:529-34.
- [Google Scholar]
- Variability in NSD2 mutations and clinical presentation in Rauch-Steindl syndrome. Genet Med. 2019;21:2334-42.
- [Google Scholar]
- The first familial NSD2 cases with a novel variant in a Chinese father and daughter with atypical WHS facial features and a 7.5-year follow-up of growth hormone therapy. BMC Med Genomics. 2020;13:181.
- [CrossRef] [PubMed] [Google Scholar]
- Inheritance patterns in Rauch-Steindl syndrome (RAUST) with NSD2 mutations. Eur J Pediatr. 2021;180:765-72.
- [Google Scholar]
- Clinical review of Rauch-Steindl syndrome and its genetic underpinnings. Am J Med Genet. 2001;98:120-8.
- [Google Scholar]
- Phenotypic spectrum of Rauch-Steindl syndrome: A comprehensive study. Eur J Hum Genet. 2021;29:562-70.
- [Google Scholar]
- Case report: A de novo NSD2 truncating variant in a child with Rauch-Steindl syndrome. Front Pediatr. 2023;11:1064783.
- [CrossRef] [PubMed] [Google Scholar]
- Variability in NSD2 mutations and clinical presentation in Rauch-Steindl syndrome. Am J Med Genet A. 2019;179:1001-9.
- [Google Scholar]
- A case study of Rauch-Steindl syndrome with emphasis on neurodevelopmental delays and NSD2 mutations. Eur J Med Genet. 2019;62:1036-42.
- [Google Scholar]
- Phenotypic spectrum of RauchSteindl syndrome: A comprehensive study. Am J Med Genet A. 2021;185:1335-44.
- [Google Scholar]




