
Translate this page into:
Kartagener’s syndrome in an adolescent male: A case report and review of literature
*Corresponding author: Thirunavukkarasu Arun Babu, Department of Pediatrics, All India Institute of Medical Sciences (AIIMS), Mangalagiri, Andhra Pradesh, India. babuarun@yahoo.com
-
Received: ,
Accepted: ,
How to cite this article: Chirag R, Rasuri A, Arun Babu T. Kartagener’s syndrome in an adolescent male: A case report and review of literature. Karnataka Paediatr J. 2024;39:102-5. doi: 10.25259/KPJ_11_2024
Abstract
Kartagener’s syndrome (KS) is a rare autosomal recessive genetic condition causing disruption to ciliary movement, leading to the triad of sinusitis, situs inversus, and bronchiectasis. Mutations in genes such as DNAI1 and DNAH5 increase susceptibility to recurrent sinopulmonary infections, infertility and errors with the left-right body orientation. A teenage boy with a decade-long history of sinusitis, chronic cough and ear infections showed bronchiectasis and situs inversus in clinical and imaging examinations. He had a neonatal intensive care unit stay for 1 month due to respiratory distress at birth, where dextrocardia was noted. Treatment with antibiotics, mucolytics, chest therapy and vaccination improved his symptoms. KS should be considered in newborns with dextrocardia and breathing problems. Genetic counselling and fertility issues should be addressed once KS is diagnosed.
Keywords
Kartagener’s syndrome
Chronic sinusitis
Bronchiectasis
Situs inversus

INTRODUCTION
Kartagener’s syndrome (KS) is a rare autosomal recessive genetic disorder affecting ciliary movement, characterised by sinusitis, situs inversus and bronchiectasis. It occurs in approximately 1 in 30,000 live births and was first described by Siewert in 1904. The clinical syndrome, including chronic sinusitis, bronchiectasis and situs inversus, was recognised by Manes Kartagener in 1933.[1] Ciliary dyskinesia was suggested as its cause by Camner et al., in 1975, while the term ‘immotile cilia syndrome’ was coined by Eliasson et al., in 1977 to highlight infertility alongside sinopulmonary infections.[2, 3] KS results from gene mutations of more than 50 genes, including DNAI1 and DNAH5, impairing ciliary motility crucial for respiratory defence, sperm motility and proper embryonic visceral orientation during embryogenesis.[4] This case report describes an adolescent boy with KS, reported due to its rarity and aims to contribute toward a greater understanding of KS.
CASE REPORT
A 13-year-old male developmentally normal adolescent, born out of a non-consanguineous marriage, presented to the paediatrics outpatient department complaining of recurrent episodes of productive cough since 3 years of age. There was a history of increased coughing when exposed to dust and shortness of breath on exertion. The child had daily symptoms of cough with shortness of breath for which he was being treated as poorly controlled bronchial asthma; however, it was mildly relieved on metered dose inhaler (Salbutamol and Formoterol). The child also had a history of suppurative otitis media of the left ear and chronic sinusitis since 3 years of age. There is no history of chest pain, haemoptysis, fever or weight loss. There was no similar history or history of atopy in the family. There was no history of tuberculosis contact. The child was born at full term by vaginal delivery with birth weight of 3 kg. The baby was admitted to the neonatal intensive care unit immediately after birth in view of respiratory distress for about a month, suspected to be meconium aspiration syndrome. A chest X-ray was done, which suggested the heart to be on the right side. General examination revealed no remarkable findings. Vitals were stable. The child was well-nourished, with anthropometry within normal limits. Respiratory examination revealed bilateral coarse crepitations and extensive wheeze all over the lung fields. Cardiovascular examination revealed heart sounds being heard clearer on the right side of the chest with no murmur.
A complete hemogram was done, which showed haemoglobin – 12.1 g/dL, total leucocyte count – 14.15 × 10*9/L, red blood cell count – 5.55 × 10*12/L, platelets – 554 × 10*6/L, mean corpuscular volume – 66.4 fl, mean corpuscular haemoglobin – 21.8 pg and mean corpuscular haemoglobin concentration – 32.8 g/dL. Differential leukocyte count showed N – 62, L – 26, M – 8 and E – 4. Serum immunoglobulin E levels showed 150.5 IU/mL (Normal range: 0 – 160 IU/mL). Sputum for acid-fast bacilli was negative. C-reactive protein and erythrocyte sedimentation rates were within normal limits. The Mantoux test was negative.
Chest X-ray showed apex and aortic arch of the heart on the right side, gastric bubble on the right side and prominent bilateral bronchovascular marking in lower lobes [Figure 1]. An electrocardiogram (ECG) was done, which suggested right axis deviation, inversion of all complexes in the lead I (inverted P wave, negative QRS, inverted T wave), upright p wave in aVL lead and an absent R wave progression and prominence of S wave in the anterior leads and low voltage in leads V4–V6 [Figure 2]. The ECG findings were normal for the right-sided chest leads. Ultrasonography abdomen revealed a liver on the left side and spleen on the right side of the abdomen, suggesting situs inversus. His pulmonary function tests suggested decreased forced vital capacity (FVC) (1.59 L or 64% predicted), decreased forced expiratory volume in one second (FEV1) (1.37 L or 65.0% predicted) and Tiffeneau-Pinelli index (FEV1/FVC) (86.1% with 98% predicted) suggestive of restriction. Echocardiography was done, which confirmed situs inversus, with no other structural heart defect. A High-resolution computed tomography scan of the lungs was performed, which showed bilateral, central, cylindrical, and varicose bronchiectatic changes with extensive mucosal plugging along with centrilobular benching nodules in bilateral lungs suggestive of infective bronchiolitis [Figures 3 and 4]. With the above-mentioned history, the primary ciliary dyskinesia clinical prediction rule (PICADAR) score[5] came to 11. Along with clinical signs and imaging studies, the diagnosis of KS was made.
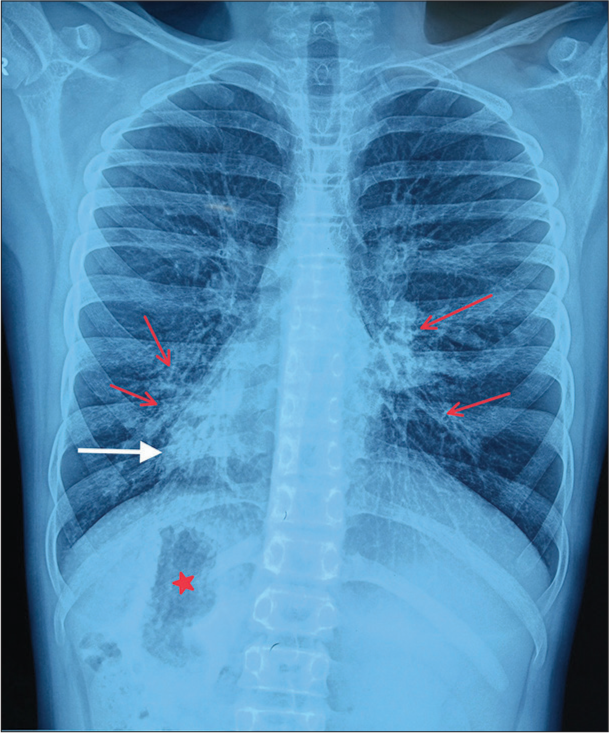
- Chest X-ray showing apex of heart on the right side (shown as white arrow), gastric bubble on the right side (shown as red star mark), bilateral bronchovascular marking prominent in lower lobes, consistent with bronchiectasis (red arrow).
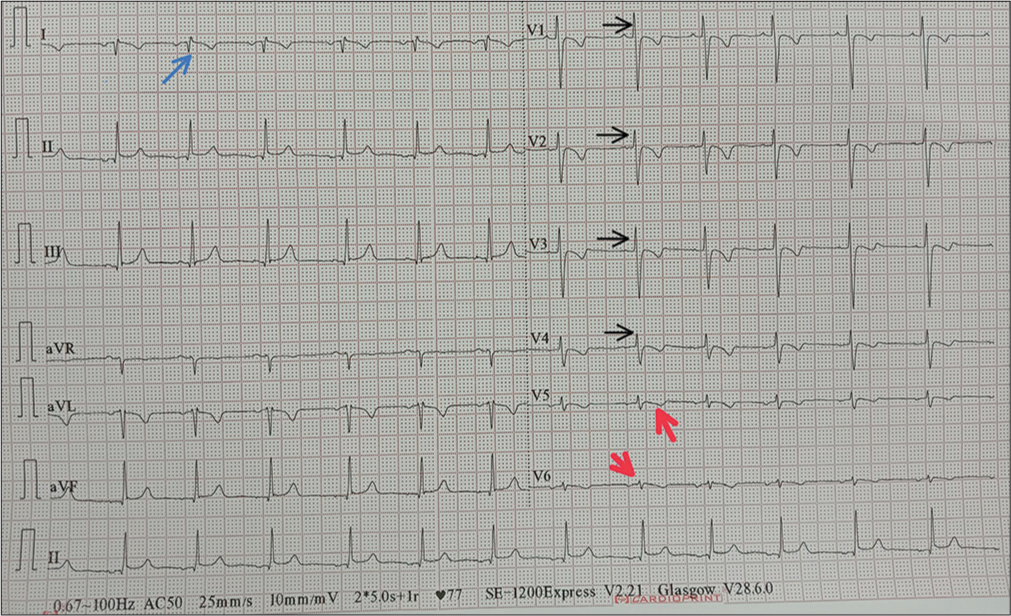
- Electrocardiogram showing right axis deviation, inversion of all complexes in lead I (inverted P wave, negative QRS, inverted T wave) shown as blue arrow, an absent R wave progression and prominence of S wave in the anterior leads (shown as black arrow) and low voltage in leads V4–V6 (shown as red arrow).
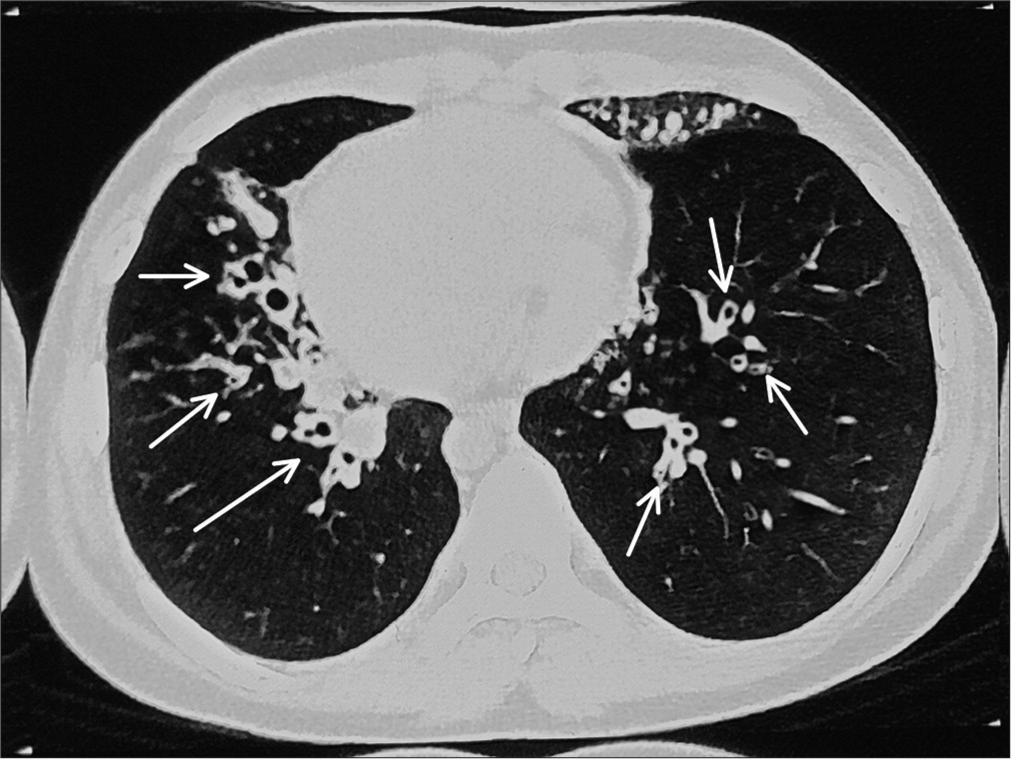
- High-resolution computed tomography showing bilateral central cylindrical and varicose bronchiectatic changes with extensive mucosal plugging (shown as white arrow) with centrilobular benching nodules in bilateral lungs suggestive of infective bronchiolitis.
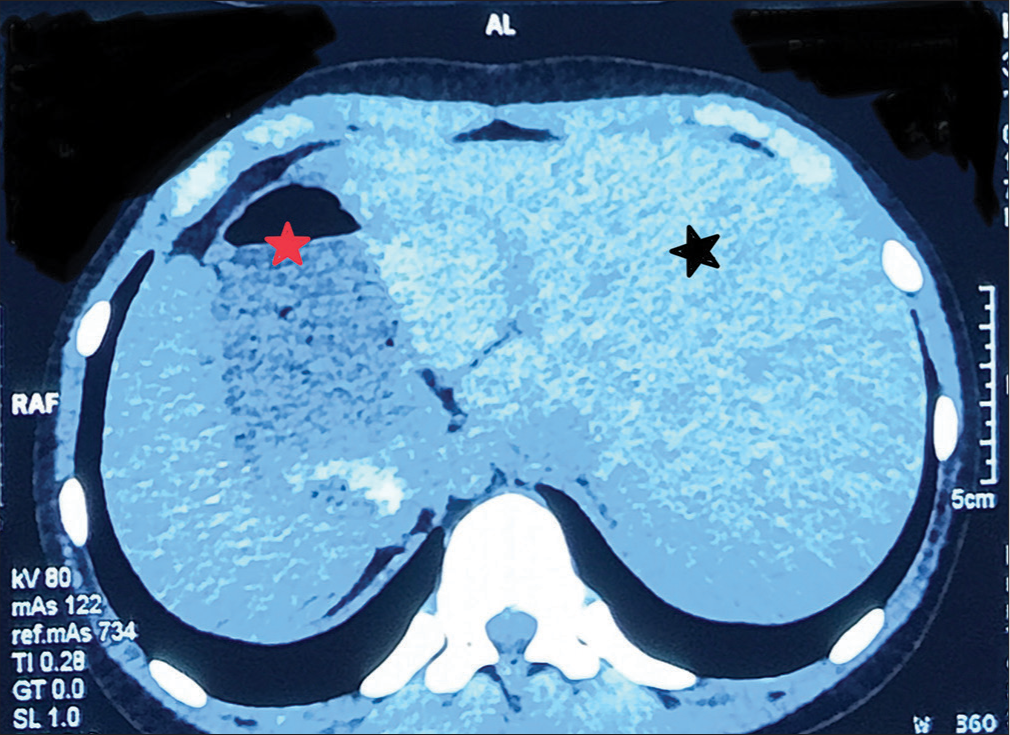
- High-resolution computed tomography showing liver on the left (black star) and stomach bubble on the right side (red star).
Antibiotics, mucolytics, inhaled bronchodilators and chest physiotherapy were advised. The child was vaccinated with influenza and pneumococcal vaccine. He was advised to avoid cough suppressants. Otorhinolaryngology consultation was taken in view of chronic suppurative otitis media of the left ear and chronic sinusitis. Pus culture revealed Staphylococcus aureus bacterial growth and the child was advised of medical management and close follow-up.
The nature, outcome and prognosis of the condition with a risk of future infertility and the importance of regular follow-up have been explained to the child’s parents.
DISCUSSION
KS is a rare disorder occurring in about 1 in 30,000 live births.[6] It falls under the category of primary ciliary dyskinesia (PCD), a congenital condition characterised by abnormalities in ciliary structure or function. This condition, which follows an autosomal recessive inheritance pattern, involves faulty coding for dynein protein, with DNAH5 and DNAI1 being the most commonly mutated genes.[7] These genetic mutations result in cilia that are misshapen, incorrect in size, or moving improperly, leading to impaired ciliary motility.[4] Normal ciliary function is crucial for proper embryonic development, respiratory health and sperm movement. Malfunctioning cilia can cause defects in the left-right body orientation, such as situs solitus or situs inversus totalis, as well as recurrent sinus and lung infections and infertility.
KS manifests as a combination of chronic sinusitis, bronchiectasis and situs inversus, though symptoms can vary among individuals. Neonates may initially experience respiratory distress, progressing to chronic cough due to bronchiectasis, unresponsive asthma, recurrent sinus infections, otitis and fertility issues such as ectopic pregnancy in females or infertility in males.[8] Diagnosis should be considered in term neonates with dextrocardia and unexplained respiratory distress or pneumonia.[9] In our case, the patient displayed the classic triad of sinopulmonary infections, situs inversus and central bronchiectasis. While initially presenting with neonatal respiratory distress and situs inversus, the diagnosis was missed. Subsequent treatment for poorly responsive asthma should have prompted consideration of ciliopathy. PICADAR score is a diagnostic predictive tool used for identifying patients who may have PCD. A full score of 14 points corresponds to a 99.80% probability of having PCD, while a score of ≥10 indicates a 92.6% probability, and a score of ≥5 indicates an 11.10% probability.[5]
Additional diagnostic tests may involve assessing nasal epithelial mucociliary function, typically measured by decreased nasal nitric oxide levels. Electron microscopy can reveal reduced ciliary beat frequency (<11 Hz/s) and absent ciliary dynein arms. Genetic studies may also be conducted to identify mutations in the DNAI1 and DNAH5 genes.[10,11] These tests have not been performed in our case due to financial limitations.
Infertility primarily results from compromised ciliary motility affecting sperm tail function and fallopian tube movement, alongside significant lung implications. In rare instances, cardiac anomalies beyond dextrocardia, such as ventricular septal defects, transposition of great arteries and pulmonary outflow defects, may also occur.[12] In addition, conditions such as pancreatic malformations, polycystic kidney disease, polysplenia and vascular malformations, although uncommon, have been documented in some cases. As a result, PCD is increasingly acknowledged as a multi-system disorder.[13]
Timely identification and consistent medical monitoring are imperative for patients with these conditions to mitigate potential complications, given the absence of a definitive cure. Supportive care for sinopulmonary issues in KS typically involves chest physiotherapy, inhaled mucolytics and antibiotic therapy. Since ciliary function is compromised, cough serves as a vital mechanism for mucus clearance; therefore, cough suppressants should be avoided. Those experiencing frequent bronchiectasis exacerbations (≥3 times/year) may require long-term and low-dose prophylactic antibiotics. Due to heightened susceptibility to upper and lower respiratory infections, vaccinations such as influenza and pneumococcal are administered. Late diagnosis of bronchiectasis is associated with a poor prognosis, even with optimal management strategies.[14]
CONCLUSION
Recognizing and considering rare conditions like Kartagener syndrome and primary ciliary dyskinesia (PCD) in the differential diagnosis of chronic respiratory infections is crucial. Early identification leads to timely treatment and improved outcomes, preventing complications such as pneumonia and bronchiectasis. Awareness among healthcare providers is key, especially in specific patient groups like neonates with dextrocardia and respiratory distress, or children with persistent wheezing unresponsive to typical asthma therapies. In these cases, considering these syndromes is essential for early diagnosis, optimal management, and enhanced quality of life.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript, and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Evidence for congenitaly nonfunctioning cilia in the tracheobronchial tract in two subjects. Am Rev Respir dis. 1975;112:807.
- [Google Scholar]
- Camner P, Afzelius BA. The immotile cilia syndrome: A congenital ciliary abnormality as an etiologic factor in chronic airway infections and male sterility N Engl J Med. 1977;. ;297:1-6.
- [CrossRef] [PubMed] [Google Scholar]
- Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol. 2003;112:518-24.
- [CrossRef] [PubMed] [Google Scholar]
- PICADAR: A diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J. 2016;47:1103-12.
- [CrossRef] [PubMed] [Google Scholar]
- Primary ciliary dyskinesia: A genome-wide linkage analysis reveals extensive locus heterogeneity. Eur J Hum Genet. 2000;8:109-18.
- [CrossRef] [PubMed] [Google Scholar]
- A rare case of Kartagener syndrome presenting with sinusitis, situs inversus, and bronchiectasis: Emphasizing early diagnosis and management strategies. Cureus. 2023;15:e41890.
- [CrossRef] [Google Scholar]
- Primary ciliary dyskinesia diagnosed by electron microscopy in one case of Kartagener syndrome. Rom J Morphol Embryol. 2014;55(2 Suppl):697-701.
- [Google Scholar]
- A case of unusual presentation of Kartagener's syndrome in a 22-year-old female patient. Cureus. 2022;14:e28119.
- [CrossRef] [Google Scholar]
- Low levels of nasal nitric oxide (NO) correlate to impaired mucociliary function in the upper airways. Acta Otolaryngol (Stockh). 1997;117:728-34.
- [CrossRef] [PubMed] [Google Scholar]
- A case of Kartagener syndrome with pulmonary hypertension. J Med Cases. 2016;7:326-80.
- [CrossRef] [Google Scholar]
- Dextrocardia and corrected transposition of the great arteries (I,D,D) in a case of Kartagener's syndrome: A unique association. Clin Cardiol. 1998;21:298-9.
- [CrossRef] [PubMed] [Google Scholar]
- Coincidence of polysplenia, Kartagener syndrome, dorsal pancreas agenesis, and polycystic kidney disease in an adult. Eurasian J Med. 2017;49:152-4.
- [CrossRef] [PubMed] [Google Scholar]
- A case of Kartagener's syndrome: Importance of early diagnosis and treatment. Indian J Hum Genet. 2013;19:266-9.
- [CrossRef] [PubMed] [Google Scholar]




