
Translate this page into:
Lumps and bumps in a young child: Hyaline fibromatosis syndrome
*Corresponding author: Viola Elvia Sequeira, Department of Dermatology, St John’s Medical College Hospital, Bengaluru, Karnataka, India. nicholesequeira@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Augustine M, Ballal S, Tirumalae R, Sequeira VE. Lumps and bumps in a young child: Hyaline fibromatosis syndrome. Karnataka Paediatr J. 2024;39:98-101. doi: 10.25259/KPJ_12_2024
Abstract
Hyaline fibromatosis syndrome (HFS) is a rare deposition disorder, wherein the skin and internal organs have unusual amounts of amorphous hyaline material. HFS can present as a milder form called Juvenile Hyaline Fibromatosis or severe Infantile Systemic Hyalinosis (ISH). Skin changes common to both the entities are skin thickening, perianal nodules, facial papules, gingival hyperplasia, hyperpigmented plaques over flexural joints and massive subcutaneous scalp tumours. Conspicuous involvement of the joints in the form of joint contractures affects patient morbidity. Furthermore, ISH involves the gastrointestinal tract characterised by protein-losing enteropathy and malabsorption. This contributes to the high mortality seen in ISH. We describe a 2-year-old male patient with grade 2 HFS who succumbed to a lower respiratory tract infection, a manifestation not typically observed in moderate HFS cases.
Keywords
Juvenile hyaline fibromatosis
Infantile hyaline fibromatosis
Hyaline fibromatosis syndrome
Joint contractures
Subcutaneous scalp tumours

INTRODUCTION
Hyaline Fibromatosis Syndrome (HFS) is a rare deposition disorder of autosomal recessive inheritance. Juvenile hyaline fibromatosis (JHF), the milder form, presents with cutaneous papules and nodules, gingival hyperplasia and variable degrees of bone involvement. However, Infantile Systemic Hyalinosis (ISH), the severe form, is distinguished by widespread hyaline deposition in various organs, particularly the gastrointestinal tract. It presents with protein-losing enteropathy and malabsorption, which, in turn, leads to recurrent infections. Most affected individuals succumb to the illness within the first 2 years of life.[1]
CASE REPORT
A 2-year-old male child, born of a second-degree consanguineous marriage and normal full-term vaginal delivery, presented with inability to extend the knees and wrist joints since 6 months of age, and asymptomatic skin lesions involving the scalp, neck and back for 1 year. There was no history of diarrhoea or recurrent respiratory tract infections. He had short stature and was severely underweight, that is <−3 Standard deviation (SD) in the World Health Organization growth chart.
Cutaneous examination showed multiple, skin-coloured tumours over the scalp, chest and back [Figures 1 and 2]. Innumerable, grouped, pink to skin-coloured, smooth, flat to dome shaped papules were noted over the face, scalp, ears, neck, chest and back [Figure 3]. Gingival tumours were also seen. Movements of the wrist and knee joints were limited, with fixed contractures over forearm and thighs. Developmental milestones were adequate for age. Systemic examination was normal. Laboratory investigations including complete blood count, liver function tests and renal function tests were within normal limits. Radiographic examination of the long bones revealed osteopenia and soft-tissue contractures. Ultrasonography of scalp showed lobulated soft-tissue intensity lesions without the involvement of underlying bone. Magnetic resonance imaging brain and fundoscopy were normal. Histopathological examination of skin lesions showed Periodic acid-Schiff (PAS)-positive, diastase resistant homogenous, eosinophilic deposits in the papillary dermis and with epidermal atrophy [Figure 4a and b]. The above features were diagnostic of HFS. The patient was referred to plastic surgery for palliative removal of the tumours and to orthopaedics for release of soft-tissue contractures. The parents were offered genetic counselling. However, genetic testing was not done due to financial constraints. Unfortunately, the child passed away 6 months later due to a lower respiratory tract infection.

- Multiple, large, firm, skin-coloured and non-tender tumours involving the scalp.
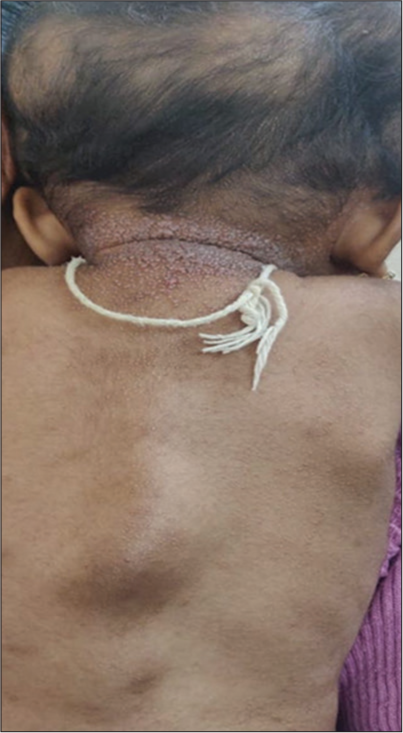
- Tumours over the spine.
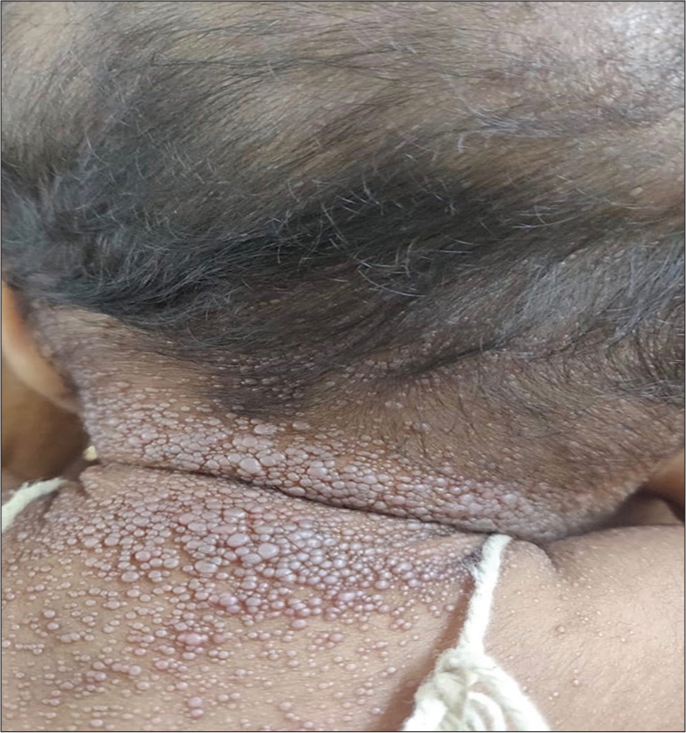
- Innumerable, grouped, pink to skin-coloured, smooth, flat to dome-shaped papules over the neck, scalp and ears.
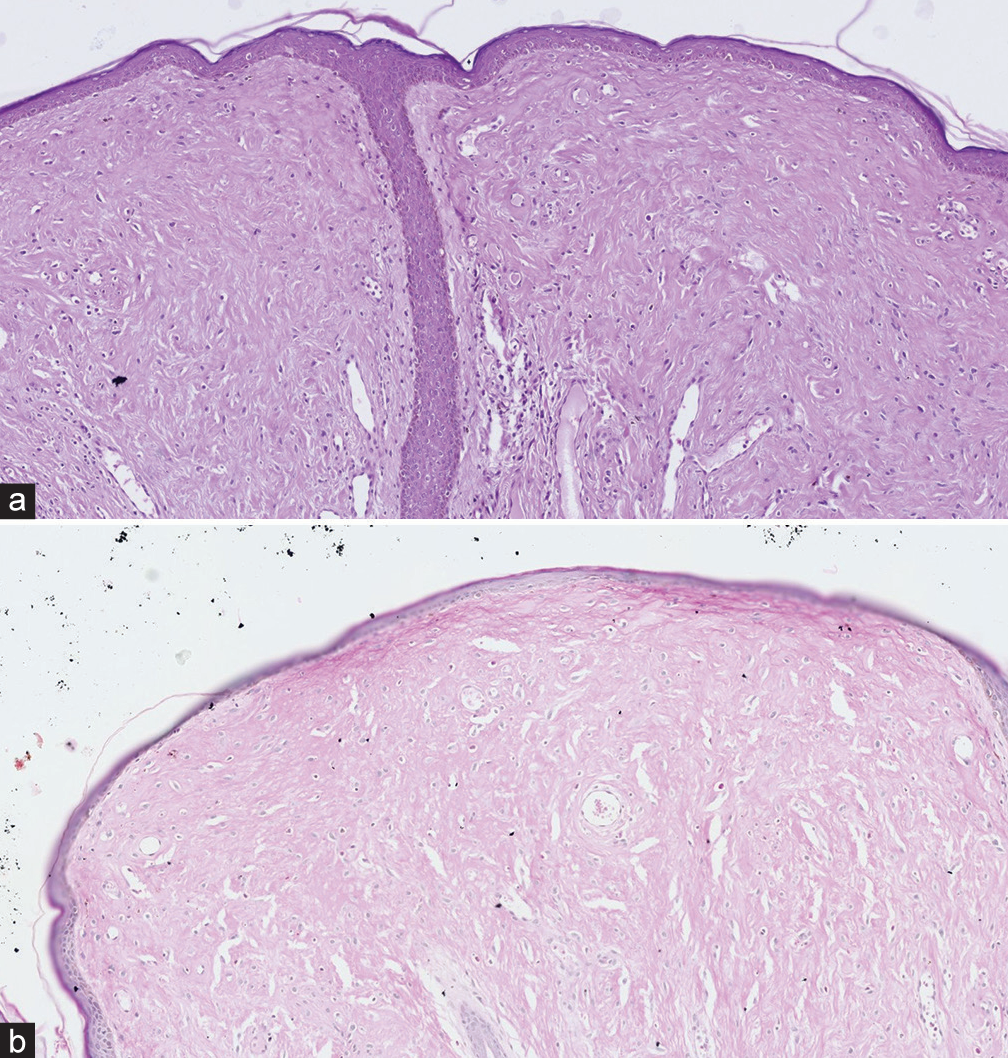
- (a) Homogenous, eosinophilic deposits in the papillary dermis, with epidermal atrophy (Hematoxylin and eosin (H&E), ×10). (b) Periodic acid-Schiff positivity in the papillary dermis (×10).
DISCUSSION
HFS is a spectrum disease that includes JHF and infantile hyaline fibromatosis (IHF). It is marked by intracellular hyaline deposits.
The incidence of the disease is not well-known. However, medical literature shows fewer than 70 documented cases of JHF and 20 cases of ISH.[2-5]
HFS is an autosomal recessive disorder caused by loss-of-function mutations in the anthrax toxin receptor 2 (ANTXR2) gene located on chromosome 4q21.21. ANTXR2 encodes for a transmembrane protein, which plays a critical role in endothelial cell morphogenesis and is essential for maintaining skin integrity. Mutations in ANTXR2, which can be homozygous or compound heterozygous, disrupt its function, leading to the abnormal deposition of hyaline material observed in HFS. According to genotype-phenotype tests, ISH is typically caused by truncating mutations and mutations affecting the extracellular domain, whereas JHF is caused by mutations in the cytoplasmic domain.[2,3]
HFS presents with a spectrum of cutaneous manifestations. Pink to pearly-white papules, plaques are seen affecting the chin, forehead, ears, posterior neck, perianal region and nasolabial folds. The scalp is characterised by large subcutaneous nodules. In addition, hyperpigmented macules and patches may be observed over bony prominences, particularly the metacarpophalangeal joints, wrists and ankles. Oral cavity involvement is seen as gingival hyperplasia, curved dental roots and tooth misalignment.[3] Radiographic investigations reveal osteopenia and periosteal reactions. Bony involvement in the form of flexion contractures affecting the proximal and distal interphalangeal joints of the hands and elbows is seen. Early adulthood is when progressive joint contractures are first noticed. In early adulthood, progressive joint contractures, often involving major joints, restrict patients to either bed or wheelchair.[3]
IHF is comparable to JHF, but the former affects the joints much more severely, resulting in joint contractures. In contrast to JHF, ISH presents with significant visceral involvement within the first few weeks or months of life. The most common feature is hyaline infiltration of the small intestine and colon, disrupting their absorptive function. This manifests clinically as malabsorption and protein-losing enteropathy, leading to diarrhoea, failure to thrive, growth failure, osteopenia and an increased susceptibility to infection. Severe joint pain and restriction cause respiratory insufficiency and immobility. By the time, a child is 2 years old, sepsis with associated renal, pulmonary and cardiac failure frequently results in death.[6]
Histologically, the epidermis appears normal, while the dermis shows characteristic hyaline deposition. This eosinophilic, PAS-positive material accumulates in both papillary and reticular dermis, within the extracellular and perivascular spaces. Spindle cell proliferation without atypia and minimal inflammation are also observed.[3]
Proposed grading system for HFS adapted from Denadai et al.[7]
Our child had skin, gingival, joint and bone involvement but did not present with protein-losing enteropathy, recurrent infections, diarrhoea and failure to thrive and was thus graded as moderate HFS (Grade 2) [Table 1]. Diagnosis in our case was based on presence of typical clinical manifestations and subsequently confirmed by histopathological analysis. Molecular testing was not done due to financial constraints.
| Feature | Grade 1 (Mild) | Grade 2 (Moderate) | Grade 3 (Severe) | Grade 4 (Lethal) |
|---|---|---|---|---|
| Skin and/or Gingival | Present (+) | Present (+) | Present (+) | Present (+) |
| Involvement | ||||
| Joint and/or Bone | Absent (−) | Present (+) | Possible (±) | Possible (±) |
| Involvement | ||||
| Internal Organ | Absent (−) | Absent (−) | Possible (±) | Present (+) |
| Involvement | ||||
| Severe Clinical | Absent (−) | Absent (−) | Absent (−) | Present (+) |
| Decompensation |
The differential diagnosis of HFS includes several disorders, such as congenital generalised fibromatosis, lipoid proteinosis and Farber disease. Farber disease can be distinguished from HFS by its neurologic involvement and lack of hyperpigmented patches. Lipoid proteinosis presents with hoarseness as the initial symptom followed by tongue enlargement, dental hypoplasia, the formation of papules around the eyelids and on the face and specific skin lesions (vesicles and crusted bullae that develop into waxy plaques) which are not seen in HFS. Multiple, or generalised, nodules made up of smooth muscle and differentiated fibroblast cells without the typical HFS skin abnormalities are the hallmarks of congenital generalised fibromatosis.[8]
At the moment, HFS lacks any definitive treatment. The sole goal of therapy is palliative. Early surgical intervention may be attempted to halt disease progression in localised lesions; however, recurrences may occur after excision.[9]
Intralesional corticosteroids may offer some benefit in managing early-stage HFS lesions. However, their effectiveness is limited in advanced lesions, which can become quite large and ulcerate, causing significant patient discomfort.[9]
Physiotherapy has little impact, although capsulotomy of the joints may have some short-term, positive effects like increased joint mobility.[10] Partial or radical gingivectomy is done for gingival overgrowth.[9]
CONCLUSION
Our patient had grade 2 HFS. Unfortunately, the patient succumbed to a lower respiratory tract infection, a manifestation not typically observed in moderate HFS cases. Given this atypical outcome, genetic testing is essential for further management and a deeper understanding of the pathogenesis of HFS.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Juvenile hyaline fibromatosis and infantile systemic hyalinosis: Divergent expressions of the same genetic defect? Indian J Dermatol Venereol Leprol. 2008;74:371-4.
- [CrossRef] [PubMed] [Google Scholar]
- Hyaline fibromatosis syndrome: Clinical update and phenotypegenotype correlations. Hum Mutat. 2018;39:1752-63.
- [CrossRef] [PubMed] [Google Scholar]
- Juvenile hyaline fibromatosis. Indian J Dermatol. 2011;56:731-3.
- [CrossRef] [PubMed] [Google Scholar]
- Juvenile hyaline fibromatosis in siblings. Indian J Dermatol Venereol Leprol. 2005;71:115-8.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile systemic hyalinosis: A fatal disorder commonly diagnosed among Arabs. Clin Exp Rheumatol. 2005;23:717-20.
- [Google Scholar]
- Infantile systemic hyalinosis. J Am Acad Dermatol. 2004;50:S61-4.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of 2 novel ANTXR2 mutations in patients with hyaline fibromatosis syndrome and proposal of a modified grading system. Am J Med Genet A. 2012;158A:732-42.
- [CrossRef] [PubMed] [Google Scholar]
- Gingival hyperplasia associated with juvenile hyaline fibromatosis: A case report and review of the literature. J Oral Maxillofac Surg. 2010;68:2604-8.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical, histologic, and ultrastructural findings in two cases of infantile systemic hyalinosis. Pediatr Dermatol. 1992;9:255-8.
- [CrossRef] [PubMed] [Google Scholar]




