
Translate this page into:
Rare phenotype of common treatable neurogenetic disorder: Wilson disease masquerading as mitochondrial disorder
*Corresponding author: Vykuntaraju K. Gowda, Department of Pediatric Neurology, Indira Gandhi Institute of Child Health, Bengaluru, Karnataka, India. drknvraju08@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Gowda VK, Roy A, Kinhal UV, Srinivasan VM. Rare phenotype of common treatable neurogenetic disorder: Wilson disease masquerading as mitochondrial disorder. Karnataka Paediatr J. doi: 10.25259/KPJ_1_2025
Abstract
Wilson disease (WD) presents with hepatic and/or neurological features. In this report, we present a child with neuro WD presenting with unusual acute neurological deficits. Seven-year-old girl born to a consanguineously married couple presented with acute-onset ataxia with weakness on the left side of the body in the past 20 days. On examination, weight, height and head circumference all were below the 3rd centile, mini-mental state score of 23/30, dysarthria, power of 3/5 left side and 4/5 on the right side, exaggerated deep tendon reflexes, extensor planters, dystonia, tremors, choreoathetosis with short stepped wide-based gait. On investigation, elevated serum glutamic-oxaloacetic transaminase (SGOT) (80 IU/l), with normal ammonia, lactate and tandem mass spectrometry. Magnetic resonance imaging brain showed features of bilateral and symmetric involvement of the globus pallidus, putamen thalamus and tegmentum of the midbrain. Slit-lamp examination showed a Kayser-Fleischer ring in both eyes. Serum ceruloplasmin was low (3.6 mg/dL) with high 24-h urinary copper excretion (250 mcg/day). Exome sequencing identified a pathogenic homozygous missense variant c.3182G>A, p.(Gly1061Glu) in exon-14 of the ATP7B gene. To conclude, WD should be suspected in a child with acute neurological manifestations such as stroke-like episodes and movement disorder, as this entity is treatable.
Keywords
Inborn errors of metabolism
Mitochondrial disorders
Stroke-like episodes
Wilson disease

INTRODUCTION
Wilson disease (WD: OMIM#277900) is an autosomal recessive metabolic disorder caused due to mutations in the ATP7B gene located on cytoband 13q14.3. ATP7B encodes adenosine triphosphatase (ATPase) copper transporting beta, which plays an important role in the transport of copper. The major systems affected are the liver and the central nervous system. WD is a great masquerader and can present with heterogeneous presentations such as Coombs negative haemolytic anaemia, psychiatric disease, autoimmune hepatitis-like presentation, Fanconi syndrome, hypoparathyroidism, cardiac involvement, recurrent miscarriages, metabolic bone disease and psychiatric disease. These atypical manifestations pose a major challenge in early diagnosis and initiation of treatment if not recognised at the initial stage of the disease.[1-3] We present one such case where WD masqueraded as stroke-like episodes mimicking a mitochondrial cytopathy.
CASE REPORT
We present a child with neuro WD presenting with unusual acute neurological deficits. Seven-year-old girl born to a consanguineously married couple presented with acute onset ataxia with weakness on the left side of the body in the past 20 days. At the onset, she started to walk by swaying on either side along with oro-facial dyskinesia with abrupt, brief movements of both upper limbs while awake state, not getting suppressed by any distraction. Weakness of the left side of the limbs gradually improved over 3–4 days. There is no past or family history of liver or neurological disease. The development of the child was normal before this presentation. On examination, weight, height and head circumference all were below the 3rd centile; mini-mental state examination score was 23/30, dysarthria, power of 3/5 left side and 4/5 on the right side, exaggerated deep tendon reflexes, extensor planters, dystonia, tremors, choreoathetosis with short stepped wide-based gait [Figure 1a].
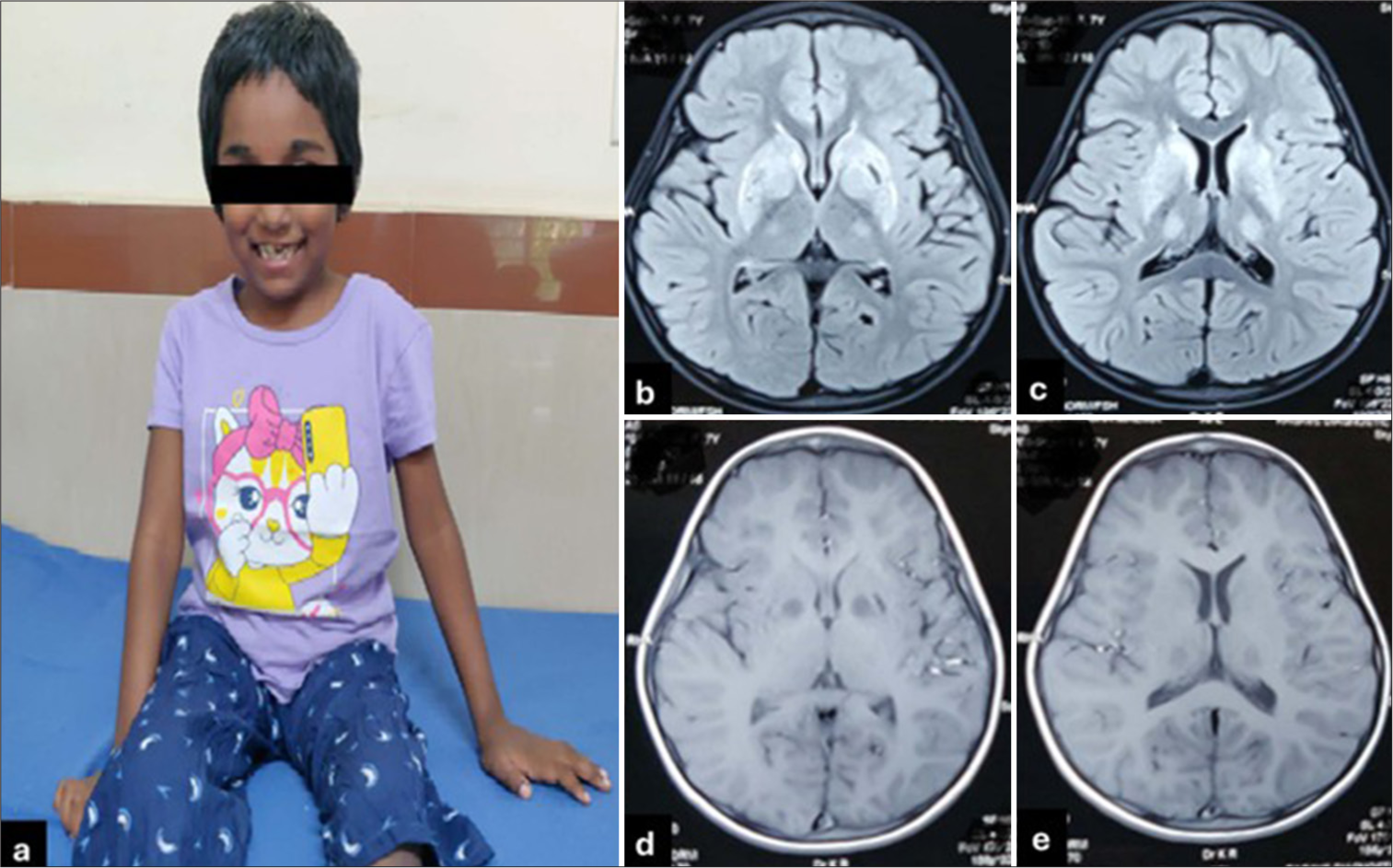
- (a) A clinical photograph of a child showing sitting with an open mouth. (b and c) T2/fluid-attenuated inversion recovery axial images showing bilateral symmetrical putamen and thalamus signal changes (T2 hyperintensity). (d and e) T1 magnetic resonance imaging images showing symmetrical hypointensity bilateral globus pallidus and thalamus.
On investigation, elevated SGOT (80 IU/l), with normal ammonia, lactate and tandem mass spectrometry. Magnetic resonance imaging brain showed features of bilateral and symmetric involvement of the globus pallidus, putamen, thalamus and tegmentum of the midbrain [Figure 1b-e and Figure 2a-f]. Slit-lamp examination showed Kayser–Fleischer rings in both eyes. Serum ceruloplasmin was low (3.6 mg/dL) with high 24-h urinary copper excretion (250 mcg/day). Exome sequencing identified a pathogenic homozygous missense variant c.3182G>A, p.(Gly1061Glu) in exon-14 of the ATP7B gene. As per American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) classification, the variant is classified as pathogenic. The variation p.(Gly1061Glu) is a well-established pathogenic variant with multiple entries on ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/312383/). Functional studies for the p.(Gly1061Glu) demonstrated that the mutation is located in the ATP binding domain and leads to retention of ATP7B protein in the endoplasmic reticulum, further leading to low levels of ATP7B protein, leading to inhibition of copper transport.
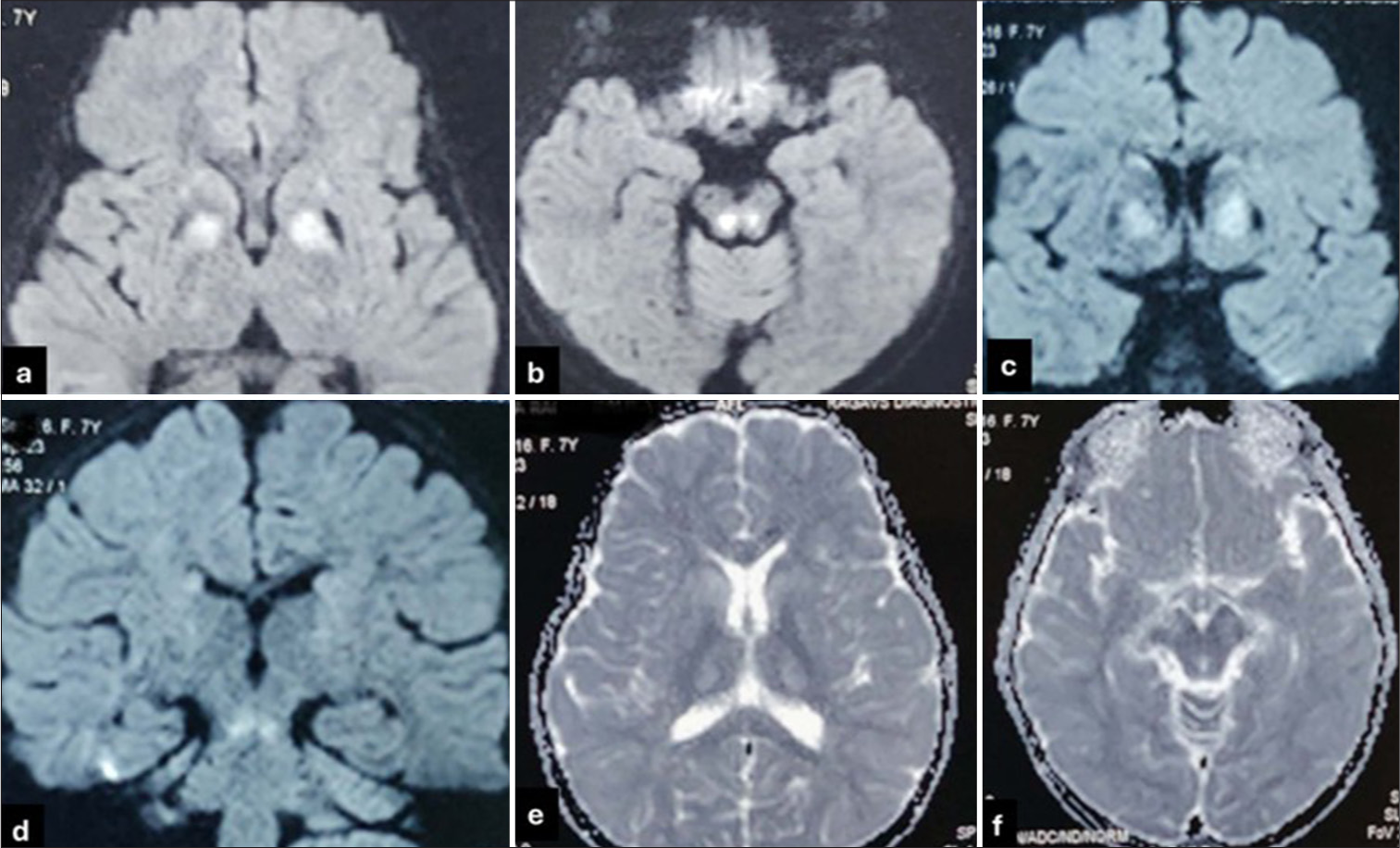
- (a and b) Diffusion-weighted images (DWI) axial images showing bilateral discrete, symmetric restricted diffusion in globus pallidus and tegmentum of midbrain. DWI axial images showing bilateral discrete, symmetric restricted diffusion in globus pallidus and tegmentum of midbrain. (c and d) DWI coronal images showing bilateral symmetrical globus pallidus and brainstem signal changes. (e and f) Apparent diffusion coefficient (ADC) shows hypodense areas in areas of DWI signal changes. ADC shows hypodense areas in areas of DWI signal changes.
DISCUSSION
The current case had features of stroke-like episodes with ataxia and movement disorder abnormalities on the background of failure to thrive. The differentials considered for stroke-like episodes were mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), other mitochondrial cytopathies due to variations in the genes MRM2-related mitochondrial DNA depletion syndrome type-17, POLG-related disease and FASTKD2-related combined oxidative phosphorylation deficiency type-44. Metabolic disorders manifesting as stroke and its variants include urea cycle disorder, especially ornithine transcarbamylase deficiency, lipoprotein dyscrasias, Fabry disease and homocysteine pathway disorders.[2,4]
Mitochondrial disorders were considered one of the major differences for the phenotype of failure to thrive, extrapyramidal manifestations, stroke-like episodes, basal ganglia involvement and acute onset of manifestations; however, points considered against the same were normal lactate and other absence of neuroimaging features of MELAS like asymmetric non-vascular territorial distribution of infarct with preferential involvement of the cortex with posterior predominance.[5] The neuroimaging features and KF rings gave us a clue about WD.
The neurological features are usually present in adults; the mean age of onset is 20–30 years, paediatric neuro WD at 7 years of age is generally considered rare. The most common features reported include movement disorders of various types, parkinsonism, tremors, dystonia, tremors of various types, dysarthria, dysphagia and seizures. Neuroimaging abnormalities are seen in 100% of drug-naive neuro WD cases, and the findings include symmetric hyperintensities involving basal ganglia, thalami, midbrain and pons with corresponding hypointense changes on T1.[6] Cystic encephalomalacia involving cortical and subcortical brain units with frontal predominance and striatal involvement has been reported in a few cases.[7,8]
Stroke and stroke-like episodes have rarely been documented in WD, with only a handful of cases reported to date. Pendlebury et al. reported a 17-year-old boy with acute-onset neurological involvement mimicking stroke-like episodes, which ultimately was diagnosed as WD due to homozygous p.(Thr766Arg) mutation in the ATP7B gene.[9] Similarly, Pan L et al. reported compound heterozygous mutations c.525dupA and c.3244-2G>A in a 14-year-old Chinese boy who presented with features of recurrent stroke-like episodes, movement disorder abnormality, basal ganglia changes and focal cortical lesion. The child was initially suspected to have a mitochondrial cytopathy: MELAS and underwent an electromyogram and muscle biopsy, which was normal. Neuroimaging gave the clue and further workup confirmed a diagnosis of WD.[10]
CONCLUSION
We report a case of neuro WD which masqueraded as mitochondrial cytopathy. The findings on neuroimaging and KF ring on slit-lamp examination gave us a clue, which was further confirmed by copper studies and molecular studies. Detailed examination, extensive literature review and a high index of suspicion for masquerades can avoid unnecessary delay in reaching a diagnosis and, finally, management of the disorder in question.
Ethical approval
Institutional Review Board has waived the ethical approval for this study.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Wilson's disease: A great masquerader. Eur Neurol. 2007;57:80-5.
- [CrossRef] [PubMed] [Google Scholar]
- Wilson disease In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews(®). Seattle, WA: University of Washington, Seattle; 1993.
- [Google Scholar]
- The wilson disease gene is a copper transporting ATPase with homology to the menkes disease gene. Nat Genet. 1993;5:344-50.
- [CrossRef] [PubMed] [Google Scholar]
- Analysis of wilson disease mutations revealed that interactions between different ATP7B mutants modify their properties. Sci Rep. 2020;10:13487.
- [CrossRef] [PubMed] [Google Scholar]
- Melas: An original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2:125-35.
- [CrossRef] [PubMed] [Google Scholar]
- Extensive striatal, cortical, and white matter brain MRI abnormalities in Wilson disease. Neurology. 2013;81:1557.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical cystic necrosis in Wilson disease. JAMA Neurol. 2016;73:350-1.
- [CrossRef] [PubMed] [Google Scholar]
- Strokelike presentation of Wilson disease with homozygosity for a novel T766R mutation. Neurology. 2004;63:1982-3.
- [CrossRef] [PubMed] [Google Scholar]
- Recurrent stroke-like episodes of Wilson disease with a novel Val176fs mutation. Neurol Sci. 2018;39:973-4.
- [CrossRef] [PubMed] [Google Scholar]




