
Translate this page into:
Situs inversus totalis in a case of Joubert syndrome
*Corresponding author: Vijay Ganesh, Department of Pediatrics, Base Hospital Delhi Cantt, New Delhi, India. vijay.bg.ganesh@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Taneja M, Ganesh V, Sangeetha B. Situs inversus totalis in a case of Joubert syndrome. Karnataka Paediatr J. 2024;39:157-60. doi: 10.25259/KPJ_36_2024
Abstract
Joubert syndrome (JS) is an uncommon genetic condition distinguished by a unique midbrain-hindbrain abnormality known as the ‘molar tooth sign (MTS)’. Classic JS is defined by three main findings: the MTS is a specific cerebellum and brain stem abnormality, hypotonia and developmental abnormalities. Over 40 causal genes have been found to date, accounting for up to 94% of cases. Individuals with JS who carry pathogenic mutations in TMEM67 are much more likely to develop liver fibrosis, whereas pathogenic variants in NPHP1, RPGRIP1L and TMEM237 are frequently associated with JS and renal involvement. Individuals with causative mutations in CEP290 or AHI1 require greater monitoring for retinal degeneration and, in the case of CEP290, chronic renal disease. These examples demonstrate how an accurate description of the range of clinical symptoms associated with abnormalities in each causal gene, particularly rare ones, would assist in improving prognosis and guide individualised care. We present a neonatal case of JS with situs inversus.
Keywords
Genetics
Joubert syndrome
Situs inversus totalis
Vermian hypoplasia

INTRODUCTION
Situs inversus (SI) represents the most extreme form, where all major organs are mirror-images from their typical positions, usually asymptomatic and most lead normal lives.[1,2] While generally well-tolerated, SI can sometimes co-occur with other developmental malformations. Vermiform hypoplasia refers to the underdevelopment of cerebellar vermis, a vital region. While the severity can vary, affected individuals may experience varying degrees of motor incoordination, gait abnormalities and speech difficulties.[3] The co-occurrence of cerebellar-vermis hypoplasia and SI in a neonate presents a unique clinical scenario.[4] A case of Joubert syndrome (JS) associated with SI in a newborn is reported [Figure 1], and the need to detect the syndrome in the neonatal period is emphasised.

- (a) Whole exome sequencing report-suggestive of Joubert syndrome. The genetic report indicating pathogenic gene involvement and (b) chromosomal microarray. Chromosomal involvement is indicated in blue colour. NA: Not applicable, OMIM- Online mendelian inheritance in man.
- Note: The red highlight indicates the genes mentioned are likely pathogenic and pathogenic variants.
CASE REPORT
This baby was a second born at term after an uneventful pregnancy, which was unnoticed till 5 months of pregnancy as the mother was under the pretext of lactational amenorrhoea and was not followed up, or antenatal follow-up was not done for the first and second trimester. The neonate was the product of a third-degree consanguineous marriage. The prenatal ultrasound identified hyperechogenic kidneys, and all other features were remarkable. The mother was not exposed to any radiation or harmful drugs during her pregnancy, with proper follow-up after the second trimester. The newborn was delivered vaginally and cried shortly after birth, with a normal Apgar at 1 and 5 min. The Apgar scores were 6 at 1 min and 8 at 5 min. Physical examination revealed a weight of 3280 g (50th percentile), a height of 48 cm (25th percentile) and a head circumference of 34 cm. The baby had a large nose bridge, anteverted nostrils and a triangular-shaped open mouth.
Soon after birth, the baby started developing respiratory distress in the form of cyanosis and an irregular pattern of respiratory (waxing and waning pattern). The baby was admitted to this level III neonatal intensive care unit for post-natal care and was given continuous positive airway pressure (CPAP) support for the initial duration. Other physical examination findings noted shifted apical impulse to the right side of the chest and liver, palpable on the left hypochondrium [Figure 2].
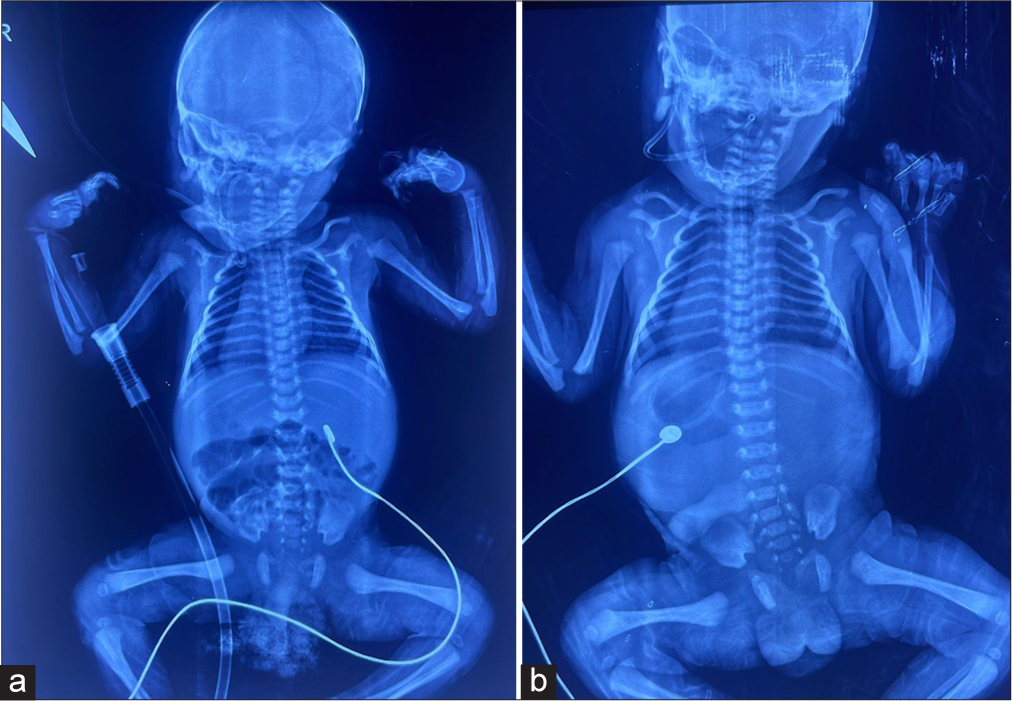
- (a) Situs inversus totalis and (b) situs inversus totalis-shift of cardiac and liver.
The initial X-ray showed SI with major organ shifts, including liver and heart. The baby had persistent distress and had to mechanically ventilate for the initial few days. The baby was treated based on an initial diagnosis of respiratory distress syndrome and early-onset newborn sepsis. After weaning off the ventilator in the 2nd week of life, respiratory assistance was reduced to CPAP. The dramatic respiratory anomaly persisted since birth, even when on assistance. The newborn was discovered to have periods of panting with a respiratory rate of up to 100 breaths/min, alternating with apnoea lasting 15–20 s without cyanosis. The baby was weaned off respiratory support by the 3rd week of life and was discharged on room air. The genetic sequencing confirmed the diagnosis of JS.
Laboratory investigations, including haemoglobin level, serum calcium level, haematocrit, phosphorus, blood urea nitrogen, creatinine, alkaline phosphatase, blood sugar, electrolytes, lactate, cerebrospinal fluid examination, acid-base values and blood and urine amino acids were normal.
X-ray and ultrasound examinations showed SI-shifted pericardium and liver. Ultrasound sonography abdomen showed increased echogenic focus in both kidneys. Further, investigations like contrast-enhanced abdominal computed tomography scans could be considered to precisely map the location of all major organs and identify any potential.
Normal cardiac anatomy and function: A 2D echocardiogram confirmed normal heart structure and function. This is reassuring, as cardiac malformations are sometimes associated with SI.
Transcranial ultrasound: Confirmed the prenatal diagnosis of vermian hypoplasia with no other associated vascular malformations [Figure 3].
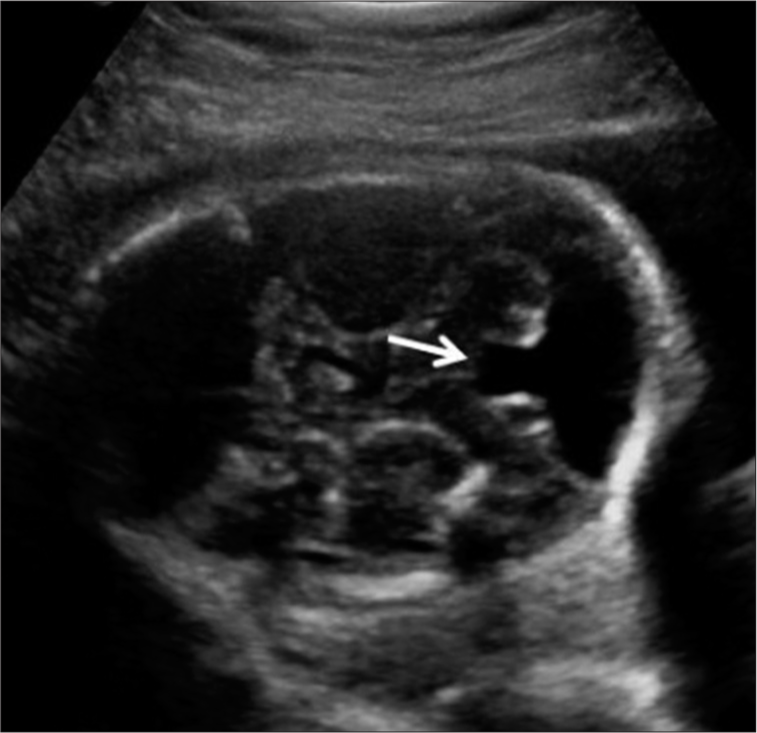
- Vermian hypoplasia transcranial ultrasound. Vermian hypoplasia- indicated through an arrow.
DISCUSSION
JS is a rare congenital neurodevelopmental primary ciliopathy with a population-based frequency of 1.7/100,000 people aged 0–19 years.[5] The clinical picture is clear from neonatal age with hypotonia, aberrant eye movements (primarily ocular motor apraxia), developmental delay and, in a subset of individuals, episodic respiratory dysregulation; subsequent clinical indicators include cerebellar ataxia and, usually, cognitive impairment.[6] Due to its complex presentation, JS is a multisystem disorder, and some clinical abnormalities may progress, complicating medical therapy.
JS can be divided into clinical categories according to related extra- central nervous system (CNS) characteristics:[6]
Purely neurological JS - (Pure JS)
JS with ocular involvement - (JS-O)
JS with renal involvement - (JS-R)
JS with oculorenal involvement - (JS-OR)
JS with hepatic involvement - (JS-H, or COACH syndrome)
JS with orofaciodigital involvement - (JS- orofaciodigital disease [OFD], or Oral-facial-digital type VI syndrome [OFDVI] syndrome)
JS with acrocallosal features
JS with Jeune asphyxiating thoracic dystrophy.
JS is typically associated with a spectrum of neurological and multi-systemic features. Here’s a few points of discussion on JS –
-
Neurological features
Molar tooth sign (MTS) – JS is distinguished by the appearance of the molar teeth sign on brain imaging, resulting from cerebellar vermis hypoplasia or aplasia, enlarged and elongated superior cerebellar peduncles and a deepened interpeduncular fossa
Cognitive Impairment – Many individuals with JS exhibit intellectual disability, ranging from mild to severe developmental delay in motor skills, language and social interaction. Early signs include developmental milestone delays, ataxia (characterised by a broad-based, unstable stride and difficulty running or climbing stairs) and intellectual handicap.[7]
-
Ocular abnormalities
Oculomotor apraxia and retinal dystrophy are noted in some individuals. This can result in impaired visual tracking and coordination.[8]
-
Renal involvement
Nephronophthisis – Renal abnormalities, particularly nephronophthisis, are common in JS – chronic kidney ds characterised by fibrosis and atrophy of the renal tubules, leading to end-stage renal disease.
Extrarenal manifestations – Additional cysts and horseshoe kidneys can be noted.[7]
Respiratory involvement Respiratory abnormalities, including episodes of abnormal breathing patterns such as hyperpnoea, apnoea and irregular breathing, are observed in a subset of individuals with JS. This can lead to respiratory distress and may require supportive interventions. As a ‘classic hallmark’ of the illness, respiratory abnormalities have been identified in all four of the siblings, which was reported first by Joubert et al.[9] It is not, however, a constant feature and reported 44% in a study by Kendall et al.,[10] 68% by Pellegrino et al.[11] and 71% by Maria et al.[12]
Five major genes (CPLANE1, CC2D2A, AHI1, CEP290 and TMEM67) accounted for approximately 6–9% of JS cases in the largest cohort yet reported; three additional genes (CSPP1, TMEM216 and INPP5E) accounted for approximately 3% of cases, and six additional genes accounted for approximately 1–2%; the remaining genes were mutated only in a minimal number of families.[3] Here, we try to emphasise that the presentation of JS with SI is not mentioned in any of the literatures, and this is one of the unique cases of JS-related disease with overlapping of SI. The gene related to this disease is CEP290 (NM_025114.4).
The association between SI totalis and vermian hypoplasia is relatively rare, and the underlying aetiology remains poorly understood. However, it is postulated that genetic factors may play a significant role, as evidenced by the consanguineous marriage of the parents in this case. Genetic counselling and further genetic testing could provide valuable insights into the inheritance pattern and potential recurrence risks for future pregnancies.
CONCLUSION
JS is a well-known autosomal recessive disorder that is uncommon. A review of the literature suggests that the disorder’s clinical symptoms do exist during the newborn period, but the right diagnosis is sometimes not obtained for several months or even years after birth. Since JS is a non-progressive illness with a wide range of presentations, the prompt diagnosis would improve the course of treatment and its final result. This study demonstrates that it is relatively possible to diagnose a new-born given the availability of genetic sequencing.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- MRI of the fetal posterior fossa. Pediatr Radiol. 2005;35:124-40.
- [CrossRef] [PubMed] [Google Scholar]
- Age and sex prevalence estimate of Joubert syndrome in Italy. Neurology. 2020;94:e797-801.
- [CrossRef] [PubMed] [Google Scholar]
- The most common comorbidities in dandy-walker syndrome patients: A systematic review of case reports. J Child Neurol. 2017;32:886-902.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis and management of the Dandy-Walker malformation: 30 years of experience. Pediatr Neurosurg. 1992;18:179-89.
- [CrossRef] [PubMed] [Google Scholar]
- Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20.
- [CrossRef] [PubMed] [Google Scholar]
- Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52:514-22.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14:583-90.
- [CrossRef] [PubMed] [Google Scholar]
- CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2010;81:104-13.
- [CrossRef] [PubMed] [Google Scholar]
- Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813-25.
- [CrossRef] [PubMed] [Google Scholar]
- Joubert syndrome: A clinico-radiological study. Neuroradiology. 1990;31:502-6.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and molecular analysis in Joubert syndrome. Am J Med Genet. 1997;72:59-62.
- [CrossRef] [Google Scholar]
- Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14:368-76.
- [CrossRef] [PubMed] [Google Scholar]




