
Translate this page into:
Type 2 caudal regression syndrome
*Corresponding author: Thirunavukkarasu Arun Babu, Department of Pediatrics, All India Institute of Medical Sciences (AIIMS), Mangalagiri, Andhra Pradesh, India. babuarun@yahoo.com
-
Received: ,
Accepted: ,
How to cite this article: Kottada G, Arun Babu T. Type 2 caudal regression syndrome. Karnataka Paediatr J. 2024;39:161-3. doi: 10.25259/KPJ_28_2024
Abstract
Caudal regression syndrome (CRS), also known as caudal dysgenesis, consists of a constellation of caudal developmental anomalies along with soft-tissue anomalies. The severity of its spectrum ranges from lumbosacral agenesis to isolated absence of coccyx. The pathophysiology of the disease is not fully known due to its rarity. In this case report, we present a case study of CRS observed in a preadolescent school-aged male child. Our objective is to contribute to advancing the literature surrounding this exceedingly rare syndrome and contextualise our findings within the broader research landscape.
Keywords
Sacral agenesis
Agenesis
Anomaly
Children

INTRODUCTION
Caudal regression syndrome (CRS), also known as sacral agenesis/caudal dysplasia, is a very rare complex congenital abnormality that affects the lower segment of the spine. It includes structural as well as neurological abnormalities of the lower spine and systemic involvement. The incidence of CRS is 1–2 in 100,000 newborns, whereas it is 1 in 350 with a maternal history of diabetes mellitus.[1] The condition was first described by Saint-Hilaire and Hohl in 1852,[2] and ‘The syndrome of caudal regression’ was first coined by Duhamel.[3] Sacral agenesis may be associated with currarino triad and homebox gene abnormalities and with neurogenic bladder dysfunction (80%).[4]
CASE REPORT
A school-aged, preadolescent male child presented to the paediatric outpatient department with chief complaints of urinary incontinence, abnormal gait and localised swelling in the right index finger, which was initially noticed by parents at 1 year of age. The baby was born to nonconsanguineous parents at term by caesarean section in view of polyhydramnios with no other maternal comorbidities. There was no history of maternal glycaemic abnormalities, exposure to teratogens or substance abuse during the antenatal period. The details of antenatal scans were unavailable. Antenatal and postnatal history was uneventful. Developmental milestones were appropriate for age. There was no family history of similar symptoms. On examination, the child had normal vitals and anthropometry appropriate for age. Musculoskeletal system examination revealed abnormal limping gait and right-sided flexion contracture of hips. The child also had a right-sided volar scaphotrapezial joint ganglion cyst. Other system examinations were unremarkable.
The anteroposterior view of the X-ray pelvis showed the absence of sacrum and coccyx with normal lumbar vertebrae and hip bones [Figure 1]. Magnetic resonance imaging (MRI) of the spine showed termination of the spinal cord at the L4 vertebral level, and the sacrum appeared to be hypoplastic with widely separated bilateral sacral ala. S2 S3 appeared small, whereas S4, S5 and coccyx were not visualised. Computed tomography (CT) pelvis with both hips was done, which showed features suggestive of partial agenesis of the sacrum (Hypoplasia of S2, S3), whereas S4, S5 and coccyx were absent. Reduced vertebral body height of the D5 vertebra (Butterfly vertebra) was noted. No evidence of meningocele was noted. Abdominal ultrasonography showed right-sided, mild hydronephrosis with bladder distension and diffuse urinary bladder wall thickening with trabeculations suggestive of chronic cystitis. The micturating cystourethrogram showed an enlarged bladder with normal urethral anatomy indicative of neurogenic bladder dysfunction [Figure 2].

- X-ray pelvis anterior-posterior view showing absent sacrum and coccyx.
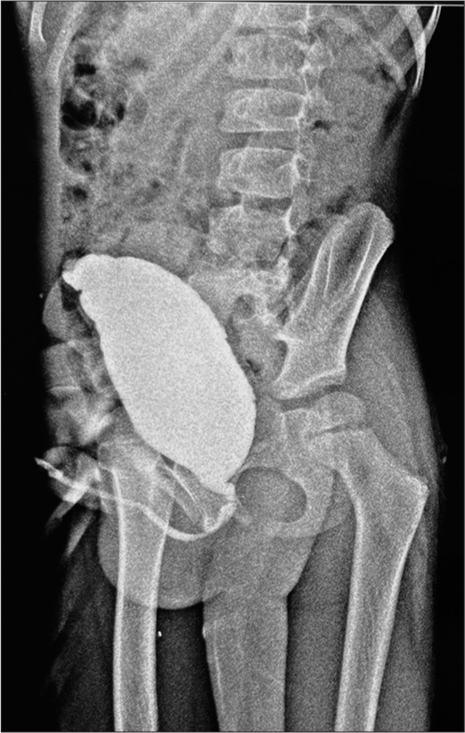
- Micturating cystourethrogram shows an enlarged bladder depicting a neurogenic bladder.
Based on the clinical presentation and radio-imaging findings, the child was diagnosed with CRS type II (with neurogenic bladder). The primary differential diagnosis of CRS includes sirenomelia, characterised by fused legs that resemble a mermaid tail, and VACTERL syndrome, with which CRS has been associated. Another relevant consideration is Currarino syndrome, characterised by a presacral mass, sacral bone defect and anorectal malformation. All these differentials were ruled out in the index case.
CRS warrants a multidisciplinary approach involving paediatric surgery, orthopaedics, neurosurgery and urogenital specialties, with early motor rehabilitation and physiotherapy as the cornerstone of treatment. In this case, there was no systemic involvement except for the neurogenic bladder. Since there was no gross systemic involvement in the child, the prognosis was expected to be relatively better. The patient was referred for a paediatric surgery consultation for urodynamic studies. The child is currently being managed conservatively and is on regular follow-up.
DISCUSSION
CRS is a rare compound congenital disorder characterised by abnormalities in the vertebral column, spinal cord, lower limb abnormalities, systemic involvement such as genitourinary abnormalities, cardiovascular and respiratory anomalies, foregut malformations, VACTERL anomalies and dysmorphic facial features.[2] Polyhydramnios is frequently associated with neural tube defects, whereas oligohydramnios is a very rare finding in CRS.[1]
The precise aetiology of this syndrome is unknown; however, factors such as maternal diabetes mellitus (insulin-dependent), vascular hypoperfusion and genetic influences are considered to be potential contributors.[5] The widely accepted theory of pathogenesis proposes that anomalies in the development of the embryonic caudal mesoderm, possibly resulting from defects in the formation of the mid-posterior axis of the mesoderm appear before the fourth week of embryogenesis, leading to skeletal malformations and organ dysplasia.[5,6] As in most reported cases, our case lacks a specific identifiable risk factor.
CRS classification, as proposed by Renshaw, delineates distinct types based on the extent and nature of anatomical involvement. This classification system provides a comprehensive framework for understanding the varied presentations of CRS. The existing literature substantiates the diagnosis of CRS prenatally by antenatal sonography scans[1] and postnatally confirmed by MRI scans.[4]
As per the published literature, common vertebral and central nervous system anomalies in CRS include hypoplastic vertebrae, vertebral fusion, hemivertebrae, butterfly vertebrae, tethered cord syndrome and spinal column malalignment (scoliosis and kyphosis) and spina bifida. In our case, the child had D5 butterfly vertebrae with reduced vertebral body height, sacrococcygeal hypoplasia with spinal cord ends at L4 vertebral level and scoliosis.
Prevalent lower limb anomalies described in the literature include talipes equinovarus, popliteal webbing, frog leg posture and short limb. Our child has an abnormal limping gait and flexion contracture of the hips. Genitourinary abnormalities described in CRS include neurogenic bladder (67% of patients of CRS),[7] vesicoureteric reflux, renal agenesis, absent bladder and transposition of external genitalia. The index case had a neurogenic bladder and bilateral hydronephrosis. An additional unique finding in our case is the presence of a right volar scaphotrapezial joint ganglion cyst, which was asymptomatic. Such association has previously been undocumented in the CRS literature. No other systemic abnormalities were found in our case.
CONCLUSION
CRS is a rare congenital condition with anomalies in the vertebral column, spinal cord, lower limbs and systemic involvement like genitourinary abnormalities. CRS can occur even in the absence of maternal gestational diabetes mellitus. Timely recognition of the condition with the delivery of informed genetic counselling is crucial for affected individuals. In all suspected cases of CRS, a complete workup of all associated systemic anomalies should be done. Further research is necessary to understand the pathophysiology of this condition.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Caudal regression syndrome: A case study with an associated review of common differential diagnoses made with antenatal sonography. J Diagn Med Sonogr. 2017;33:130-3.
- [CrossRef] [Google Scholar]
- Caudal regression syndrome: Postnatal radiological diagnosis with literature review of 83 cases. Radiol Case Rep. 2022;17:4636-41.
- [CrossRef] [PubMed] [Google Scholar]
- From the mermaid to anal imperforation: The syndrome of caudal regression. Arch Dis Child. 1961;36:152-5.
- [CrossRef] [PubMed] [Google Scholar]
- Sacral agenesis and neurogenic bladder: Long-term outcomes of bladder and kidney function. J Pediatr Urol. 2016;12:158.e1-7.
- [CrossRef] [PubMed] [Google Scholar]
- Prenatal diagnosed caudal regression syndrome. Open J Obstet Gynecol. 2013;3:227-31.
- [CrossRef] [Google Scholar]
- Sirenomelia Pathological features antenatal ultrasonographic clues, and a review of current embryogenic theories. Hum Reprod Update. 1999;5:82-6.
- [CrossRef] [PubMed] [Google Scholar]
- Treatment of patients with caudal regression syndrome: a systematic review of the literature. Russ J Spine Surg (Khirurgiya Pozvonochnika). 2023;20:21-31.
- [CrossRef] [Google Scholar]




