
Translate this page into:
Unlocking the mystery: Methylmalonic aciduria masquerading as Guillain–Barre syndrome

*Corresponding author: Vykuntaraju K. Gowda, Department of Pediatric Neurology, Indira Gandhi Institute of Child Health, Bengaluru, Karnataka, India. drknvraju08@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Gowda VK, Reddy SC, Pulikkal N, Nayyer A. Unlocking the mystery: Methylmalonic aciduria masquerading as Guillain– Barre syndrome. Karnataka Paediatr J. doi: 10.25259/KPJ_48_2024
Abstract
Methylmalonic acidemia (MMA) is a rare genetic disorder characterised by impaired amino acid metabolism. MMA presents with diverse, age-dependent symptoms, affecting multiple organ systems, especially the central nervous system. Its clinical manifestations lack a single defining characteristic, making diagnosis challenging. We report a unique case of a 1-year-old male child presenting as Guillain–Barre Syndrome, on workup, was diagnosed with MMA through laboratory investigations and genetic analysis. Prompt diagnosis and management improved patient outcomes.
Keywords
Guillain–Barre syndrome
Hyperammonemia
Metabolic acidosis
Methylmalonic acidemia
Neurological symptoms

INTRODUCTION
Methylmalonic acidemia (MMA), also known as methylmalonic aciduria, is an organic acidemia that is inherited in an autosomal recessive fashion. The clinical manifestations of MMA are diverse and non-specific, and the onset of the manifestations ranges from the neonatal period to adulthood.[1] The purpose of this case report is to highlight the rare presentation of MMA as Guillain–Barre syndrome (GBS), emphasising the importance of early diagnosis and prompt treatment.
CASE REPORT
A 1-year-old male child, born to second-degree consanguineous parents, presented to the casualty with a 1-day history of generalised weakness and fast breathing. The child’s developmental milestones were normal until the onset of symptoms. On physical examination, the child appeared sick and lethargic, with a Glasgow coma scale (GCS) score of 9/15. Vital signs revealed tachypnoea, oxygen saturation of 97% on room air, pulse rate of 152 bpm and blood pressure of 90/60 mmHg. Central nervous system examination revealed hypotonia, power 1/5 in both upper and lower limbs and areflexia. Respiratory system examination showed tachypnoea without retractions, bilateral normal vesicular breath sounds and no added sounds.
Laboratory evaluation revealed high anion gap metabolic acidosis with a pH of 7.25, partial pressure of carbon dioxide of 24.4, partial pressure of oxygen of 157, bicarbonate of 13, base excess of -15 and lactate of 18. In addition, electrolyte imbalance was noted, with sodium levels at 133 mEq/L and chloride levels at 103 mEq/L, ammonia levels at 113 micromol/L with urine ketone bodies at 3+ suggesting ketosis. Homocysteine levels were normal at 9 mg/dL. Complete blood count showed haemoglobin at 11.9 g/dL, total count at 12,210/uL, neutrophils at 42%, lymphocytes at 49 and platelets at 4.49 lak/uL. C-reactive protein was 1.2 mg/L, and erythrocyte sedimentation rate was 7 mm/1st hr. Serum creatine phosphokinase was normal. Nerve conduction studies in both lower and lower limbs were normal. In the background of consanguineous marriage, very acute onset of weakness, fast breathing and high anion gap metabolic acidosis are considered metabolic conditions with ketosis like organic aciduria.
MRI of the brain revealed bilateral globus pallidi hyperintensities on fluid-attenuated inversion recovery sequences [Figure 1a] and restricted diffusion on diffusion-weighted imaging sequences [Figure 1b]. Tandem mass spectroscopy showed mild elevation of C3 (propionyl carnitine) and 3-hydroxybutyryl carnitine, with an elevated acylcarnitines/total carnitines ratio of 0.72 (normal 0.10– 0.70). Urine organic acid (gas chromatography–mass spectrometry) analysis revealed mild-to-moderate elevation of methylmalonic acid, methyl citric acid, 3-methylglutaconic acid, acetoacetic acid, glycerol and tyrosine metabolite, 4-hydroxy phenyl lactic acid. Based on these findings, a diagnosis of methylmalonic aciduria was made. The child was started on a specific protein diet and levocarnitine supplementation. Following treatment, the child showed significant clinical outcomes. Whole-exome sequencing revealed a homozygous pathogenic variant c.433C>T, p.(Arg145Ter), in exon-2 of the MMAA gene, confirming the diagnosis of MMA of the cobalamin A complementation type.
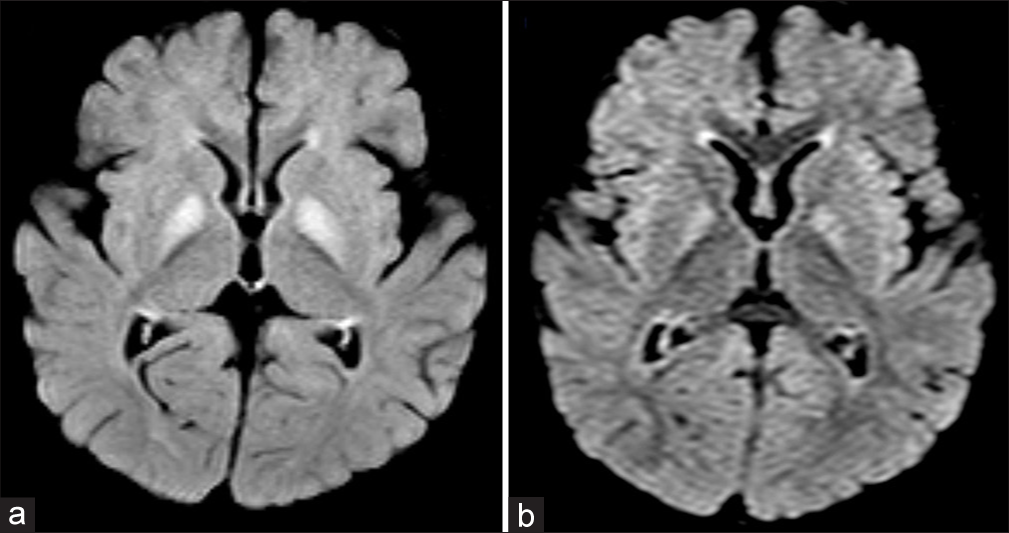
- (a) Axial fluid-attenuated inversion recovery image shows hyperintensity in bilateral globus pallidi with mild swelling. (b) Axial diffusion-weighted image shows mild hyperintensity suggestive of restricted diffusion.
DISCUSSION
MMA is the most common type of organic academia.[2] The main clinical manifestations of MMA include feeding difficulty, intellectual disability, ataxia, abnormal muscle tone, seizures and lethargy.[3] Biochemical analysis may detect abnormalities such as hyperammonemia, metabolic acidosis, hypoglycaemia and anaemia.[4] Elevated methylmalonic acid, together with 3-hydroxy propionate and the presence of 2-methyl citrate, confirm the diagnosis of MMA.[1,5] Determining the specific genetic mutation is essential for the confirmation of diagnosis, tailored management strategies and accurate genetic counselling for families.[1,5]
The underlying pathophysiological mechanism in MMA involves the inhibition of succinate dehydrogenase by elevated levels of methylmalonic acid, disrupting mitochondrial glucose oxidation. The accumulation of toxic organic acid metabolites, including methylmalonic acid, propionic acid and 2 -methylcitric acid, leads to mitochondrial dysfunction and ultimately triggers neuronal cell apoptosis.[6] In MMA with homocystinuria, impaired methionine synthase activity disrupts methylation processes, leading to homocysteine accumulation and decreased S-adenosylmethionine levels. This deficiency is linked to dysmyelination, compromising the integrity of the myelin sheath.[7] As the myelin sheath plays a crucial role in neuronal protection and facilitation of nerve impulse transmission, dysmyelination can impede nerve conduction, culminating in neuronal damage and associated neurological sequelae. In this report, we describe a 1-year 1-day-old male child who presented with GBS-like symptoms, subsequently diagnosed with MMA, which signifies the importance of considering metabolic disorders in the differential diagnosis of neurological conditions.
CONCLUSION
MMA should be considered whenever a child presents with weakness and tachyphemia in addition to other causes of acute flaccid paralysis like GBS.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Methylmalonic acidemia: Neurodevelopment and neuroimaging. Front Neurosci. 2023;17:1110942.
- [CrossRef] [PubMed] [Google Scholar]
- Toxic metabolites and inborn errors of amino acid metabolism: What one informs about the other. Metabolites. 2022;12:527.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis and treatment of methylmalonic acidemia in 14 cases. Zhonghua Er Ke Za Zhi. 2004;42:581-4.
- [Google Scholar]
- Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130.
- [CrossRef] [PubMed] [Google Scholar]
- Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J Inherit Metab Dis. 2021;44:566-92.
- [CrossRef] [PubMed] [Google Scholar]
- Brain damage in methylmalonic aciduria: 2-Methylcitrate induces cerebral ammonium accumulation and apoptosis in 3D organotypic brain cell cultures. Orphanet J Rare Dis. 2013;8:4.
- [CrossRef] [PubMed] [Google Scholar]
- Early-onset combined methylmalonic aciduria and homocystinuria: Neuroradiologic findings. AJNR Am J Neuroradiol. 2001;22:554-63.
- [Google Scholar]




