
Translate this page into:
Vitamin responsive conditions in pediatric neurology
*Corresponding author: Naveen Sankhyan, Pediatric Neurology Unit, Department of Pediatrics, Advanced Pediatrics Centre, PGIMER, Chandigarh, India. drnsankhyan@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Bhagwat C, Sankhyan N. Vitamin responsive conditions in pediatric neurology. Karnataka Paediatr J 2021;36(1):42-8.
Abstract
Vitamin responsive conditions can be either due to inherited defects in the metabolic pathways resulting in vitamin dependency or due to acquired deficiency states. Due to widespread malnutrition and predominantly vegetarian population in India, vitamin deficiency state is quite common and early identification is essential. Inherited defects, if treated earlier, lead to reduced morbidity and mortality and improvement in long-term neurocognitive outcomes. Various vitamin responsive conditions in pediatric neurology shall be discussed in this review. Infantile presentation of thiamine deficiency results in beriberi, and in adults, it leads to Wernicke’s encephalopathy and Korsakoff psychosis. Biotin thiamine-responsive basal ganglia disease is a defect of thiamine transporter 2, which leads to neuroregression and characteristic neuroimaging features of basal ganglia involvement, it responds to high doses of biotin and thiamine. Riboflavin is an enzyme involved in mitochondrial energy synthesis and is supplemented in various mitochondrial metabolic conditions. Brown-Vialetto-Van Laere syndrome is progressive pontobulbar palsy caused by defect in riboflavin transporters responsive to high doses of riboflavin. Pyridoxine responsive epilepsy presents with pharmacoresistant seizures in neonatal or early infantile age, biotinidase deficiency also presents with similar neurological manifestations, but typical cutaneous symptoms of rash and seborrheic dermatitis also occur. Both are epileptic encephalopathies and any infant presenting with epilepsy not responding to conventional AEDs must be given a trial of pyridoxine, biotin, and folinic acid. Vitamin B12 responsive conditions can include deficiency states, such as those manifesting with peripheral neuropathy and the syndrome of infantile tremor syndrome (developmental delay or regression, tremors, and megaloblastic anemia) as well as inherited disorders of homocysteine and cobalamin metabolism. These disorders are differentiated on the basis of clinical phenotype and laboratory parameters (serum B12, homocysteine levels, methylmalonic acid levels, etc.). Infantile tremor syndrome responds drastically to mega doses of Vitamin B12 and other multivitamins. Vitamin E deficiency causes ataxia with Vitamin E deficiency, other vitamins which can neurological symptoms include Vitamin C (pseudoparalysis) and Vitamin K (central nervous system bleeds). It is imperative for a practicing pediatrician to be well versed with these conditions, as these are potentially treatable conditions.
Keywords
Vitamins
Vitamin responsive epilepsies
Vitamin B12
Thiamine
Biotin

INTRODUCTION
Vitamins are naturally occurring food substances that are essential for the proper functioning of the human body in small quantities. Most of the time, they act as cofactors for various enzymatic reactions. Vitamin responsive conditions can be either due to inherited defects in the metabolic pathways resulting in vitamin dependency or due to acquired deficiency states. The nervous system, being one of the highly metabolically active systems of the human body, shows early and more severe manifestations of vitamin responsive conditions. In this review, we shall briefly discuss the various vitamin responsive conditions in pediatric neurology. A host of other disorders both acquired and inherited respond to vitamins and cofactors and the reader is referred to some good recent articles on this subject.[1]
THIAMINE (B1)
Thiamine is a water-soluble, sulfur-containing vitamin involved in key reactions of mitochondrial energy synthesis. Pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase, and branched-chain alpha-keto acid dehydrogenase are the three enzymes for which thiamine is required as a cofactor. Deficiency usually occurs in malnutrition and malabsorptive states. Infantile presentation of thiamine deficiency results in beriberi, which is of two types. In dry beriberi, there is length-dependent sensorimotor peripheral neuropathy without cardiac involvement. Wet beriberi is characterized by neuropathy as well as cardiac involvement in the form of systolic dysfunction and cardiac failure. Wernicke’s encephalopathy and Korsakoff ’s psychosis are commonly seen in alcohol-dependent adults. In children, gastrointestinal surgical procedures, recurrent vomiting, chronic diarrhea, cancer and chemotherapy, systemic diseases, drugs, and malnutrition can result in manifestations like Wernicke’s encephalopathy. The classic triad consists of encephalopathy, oculomotor dysfunction, and gait ataxia but is not seen in all patients. Manifestation such as these in young particularly malnourished children after an episode of vomiting, diarrhea should prompt its consideration.[2]
Common magnetic resonance findings include symmetric T2 hyperintensities in dorsal medial thalamus, mammillary bodies, brain stem, and basal ganglia structures.[3] Treatment with thiamine (infants IV 25–50 mg followed by 10 mg IM for a week and 3–5 mg once a day orally for 6 weeks; children 10 mg IM/IV for the 1st week, then 3–5 mg orally for 6 weeks; and adolescents 100 mg IM/IV once a day for 1 week then 10 mg orally once a day)[4-6] should be instituted immediately when the diagnosis is suspected.[7]
BIOTIN THIAMINE-RESPONSIVE BASAL GANGLIA DISEASE
Most commonly reported from Saudi Arabia, this disease is known by several names such as “Thiamine metabolism dysfunction syndrome Type 2,” “SCL19A3 gene defect,” and “biotin-responsive basal ganglia disease” “biotin thiamine-responsive basal ganglia disease.” The primary defect is in the thiamine transporter 2, encoded by the autosomal recessive SLC19A3 gene. This disease usually presents in childhood with an episode of neuroregression following a non-specific trigger such as febrile viral illness or trauma. Clinical manifestations include subacute encephalopathy, seizures, cranial nerve palsies, extrapyramidal symptoms, and quadriparesis. Rarely, infantile Leigh like the picture, as well as adult Wernicke’s encephalopathy, has also been described to be associated with this disease. MRI picture during the acute episode includes vasogenic edema in the basal ganglia region (caudate and putamen), as well as signal changes in the thalamus and white matter, later on, atrophy, which may ensue. Treatment includes high doses of biotin and thiamine (5–10 mg/kg of thiamine and 10–40 mg/kg of biotin) in the acute stages and lower doses of thiamine in the chronic stage. Prognosis depends on the age of onset, rapidity of treatment, and the clinical severity.[8]
RIBOFLAVIN (B2)
Riboflavin or B2 is a water-soluble vitamin predominantly involved in various oxidation-reduction reactions as flavin adenine dinucleotide or flavin mononucleotide. It plays an important role in the citric acid cycle, electron transport chain, beta-oxidation, and degradation of amino acids. Deficiency states include glossitis, cheilitis, seborrheic dermatitis, anemia, and axonal variety of sensory-motor neuropathy.[9] Riboflavin being an important component of the electron transport chain, it is hypothesized that riboflavin supplementation reduces cell death and necrosis in mitochondrial disorders. Various disorders potentially responsive to riboflavin include multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, migraine, multiple acyl dehydrogenase deficiency, mitochondrial complex deficiencies, glutaric aciduria, Leber’s hereditary optic neuropathy, Alpers syndrome, and Kearns-Sayre syndrome.[10]
BROWN-VIALETTO-VAN LAERE (BVVL) SYNDROME
Mutations in the riboflavin transporter genes SLC52A2 and SLC52A3, which encode riboflavin transporters, cause autosomal recessive inherited neurodegenerative disorders BVVL and Fazio Londe (FL) syndrome. This syndrome is characterized by progressive pontobulbar palsy (ataxia secondary to axonal sensorimotor neuropathy and weakness of upper limbs), nystagmus, sensorineural hearing loss (not in FL syndrome), and respiratory insufficiency. Brain MRI may be normal or may demonstrate brainstem changes. Significant improvements have been reported to occur following supplementation with high-dose riboflavin.[11] High-dose riboflavin is reserved for the treatment of metabolic defects in which riboflavin is a cofactor. These include a host of mitochondrial disorders, glutaric aciduria, and patients with mutations in the riboflavin transporter genes.
PYRIDOXINE
Pyridoxine or Vitamin B6 is a cofactor for multiple enzymatic reactions and has an important role in dopamine synthesis, serotonin synthesis, homocysteine metabolism, and conversion of glutamate to glutamate and gamma-aminobutyric acid. Pyridoxine deficiency may manifest as neuropsychiatric symptoms of anxiety and depression, sensorimotor polyneuropathy, dermatological manifestations such as seborrheic dermatitis, glossitis, cheilosis, and hematological manifestations like sideroblastic anemia. Pyridoxine deficiency may be caused by malnutrition, malabsorptive states, drugs such as isoniazid, and anti-seizure medications (phenytoin and carbamazepine). Isoniazid therapy causes a neuropathy that is reversible with pyridoxine therapy.
PYRIDOXINE-DEPENDENT EPILEPSY (PDE)
This disorder commonly presents with neonatal-onset epileptic encephalopathy unresponsive to AEDs but responsive to pyridoxine. The defect is in the lysine metabolism due to autosomal recessively inherited mutations in the ALDH7A1 gene, which encodes antiquitin or a-aminoadipic semialdehyde (aAASA) dehydrogenase. Deficiency of this enzyme results in the accumulation of aAASA/L-D1-piperidine-6 carboxylate (P6C) in the body fluids, which inactivates pyridoxal phosphate (PLP). Clinical manifestations include excessive intrauterine movements, which are actually intrauterine seizures, neonatal seizures, gastrointestinal symptoms, and developmental delay. Presentations beyond early infancy are rare, although they may occur. Neuroimaging may be normal or may show corpus callosum anomalies, ventriculomegaly, delayed myelination or hypoplasias, etc. Diagnosis is made by elevated levels of alpha-aminoadipic semialdehyde, P6C, and pipecolic acid in urine and cerebrospinal fluid (CSF), although genetic diagnosis is confirmatory. PDE is a treatable cause of epilepsy; hence’ any neonate presenting with unexplained refractory seizures should be given a trial of Vitamin B6. In a seizing infant, 100 mg of pyridoxine given intravenous results in clinical as well as an electrographic response; however, every infant should be given a trial of oral supplementation even if there is no response to the intravenous trial. The risk of side effects like apnea should be kept in mind while administering pyridoxine. The usual dose is 15–30 mg/ kg, long-term poor outcome in the form of developmental delay and neurological sequelae occurs even with cessation of seizures. Prophylactic treatment with pyridoxine has been advised for in utero treatment of suspected or proven PDE children. Some patients of PDE respond not only to pyridoxine but also to folinic acid, as both of these disorders are allelic. PNPO deficiency, which occurs due to a defect in pyridoxal 5-phosphate oxidase, responds to PLP (pyridoxal 5-phosphate) rather than to pyridoxine; this entity is known as pyridoxal 5-phosphate responsive epilepsy. Potential treatment options also include lysine restriction and arginine supplementation, which have a possible role in improving long-term neurodevelopmental outcomes, although the evidence currently is limited.[12,13]
BIOTIN
Biotin or Vitamin B7 is a cofactor for pyruvate carboxylase, propionyl-Coa carboxylase, beta-methylcrotonyl-Coa carboxylase, and two isoenzymes of acetyl-Coa carboxylase. It is involved in the key reactions of gluconeogenesis, fatty acid synthesis, and amino acid catabolism. Pathogenesis of the central nervous system (CNS) involvement is unknown, and as such dietary deficiency is rare. Two disorders that result in biotin deficiency are multiple carboxylase deficiency and biotinidase deficiency.
BIOTINIDASE DEFICIENCY
Another form of vitamin responsive condition is biotinidase deficiency. Biotinidase is an enzyme that recycles biotin in the human body. Biotinidase deficiency is an autosomal recessive inherited disease resulting in two types, profound deficiency (1–10% of enzyme activity) and the partial deficiency (10– 30% of enzyme activity). Affected children present between neonatal age to early childhood with neurocutaneous features such as seizures (generalized, partial, spasms, and myoclonic), rash, seborrheic dermatitis, hypotonia, developmental delay, and vision and hearing impairment. Laboratory studies show elevated CSF and blood lactate and pyruvate levels, organic acidemia, and urinary excretion of 3-hydroxyisovaleric acid. EEG may show severe abnormalities such as burst suppression or multifocal spikes [Figure 1]. Neuroimaging is non-specific; delayed myelination and cortical atrophy may be present. This disease responds to pharmacologic doses of biotin (5–10 mg/day), and seizures have been known to become passive by 24–48 h of administration of biotin. Recently, screening for biotinidase deficiency has been advocated in multiple neonatal screening programs, as the prompt administration of biotin improves the long-term neurocognitive outcomes.[14]
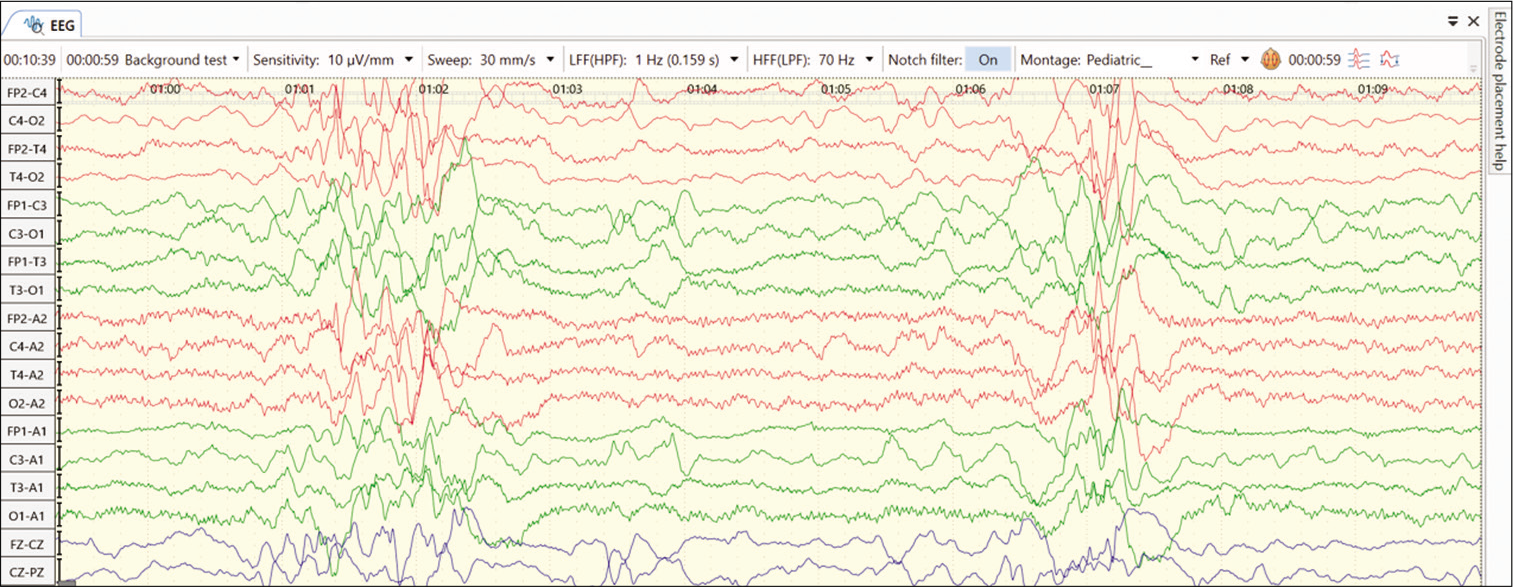
- EEG abnormality with features like hypsarrhythmia in an infant with biotinidase deficiency.
VITAMIN B12 AND FOLATE
Vitamin B12 and folate are involved in various important metabolic reactions and have protean manifestations of their deficiency and dependency states. Response to these cofactors is seen not only in the disease of a defect in their metabolic pathway but also in related other metabolic conditions. These disorders are complex; however, a brief review shall be done here, highlighting important diseases.
VITAMIN B12 METABOLISM
Vitamin B12 is found in non-vegetarian sources such as meat, milk, and eggs. Vegetarian diets are deficient in Vitamin B12. Vitamin B12 bound to food sources is released in the stomach by peptides, and it binds to haptocorrin. In the duodenum, the Vitamin B12-haptocorrin complex is broken, and Vitamin B12 is bound to the intrinsic factor; this is the complex which is absorbed in the distal ileum. In the blood circulation, B12 is bound to haptocorrin and transcobalamin; transcobalamin is the cellular delivery protein.
Vitamin B12 acts as a cofactor for two enzymes, methionine synthase, which converts homocysteine to methionine, and methylmalonyl-CoA mutase, which converts methylmalonylCoA to succinyl-CoA. The deficiency of Vitamin B12 results in the involvement of two major organ systems, hematologic, resulting in megaloblastic anemia and neurologic, as B12 is essential for the formation and maintenance of normal myelination.
Dietary deficiency of Vitamin B12 occurs commonly in adults due to pernicious anemia; however, this cause is rare in children. Children commonly suffer due to causes such as increased metabolic demands, malabsorptive states, and nutritional deficiencies. Vitamin B12 deficiency manifests as megaloblastic anemia, hypercellular marrow, pancytopenia, peripheral neuropathy, myelopathy, optic atrophy, and CNS symptoms.[15]
INFANTILE TREMOR SYNDROME (ITS)/ NEUROCUTANEOUS SYNDROME OF VITAMIN B-12 DEFICIENCY (NIB SYNDROME)
This disorder is usually seen in infants of vegetarian mothers who are themselves deficient in B12. The usual age of presentation is 6–12 months; however, this disorder can present as developmental delay from early infancy to later in the 2nd year of life. Symptoms include the triad of developmental delay or regression, tremors, and megaloblastic anemia. The affected infant has a characteristic phenotype; predominantly breastfed, they become progressively listless and apathetic, lose interest in surroundings, develop a bleating cry, chubby appearance, sparse, thin lustreless hair, and hyperpigmentation in limbs [Figures 2 and 3]. Tremors though characteristic are not seen in majority of the children. It has been shown that tremors predict poorer long-term developmental outcomes and hence indicate a more severe and prolonged deficiency of Vitamin B-12.[16] A new terminology has been suggested for this syndrome “NIB syndrome” highlighting the central role of Vitamin B-12 deficiency in its occurrence.[17] It is hypothesized that B12 is essential for normal maturation and myelination in the human brain, and hence, its deficiency results in these symptoms. MRI shows variable changes including atrophy, delayed myelination, as well as signal changes over the basal ganglia in severe cases.[18] Serum Vitamin B12 assays have low sensitivity and specificity as the major form of B12 in the serum is bound to haptocorrin; however, B12, which is available at the tissue level, is the one bound to transcobalamin. Homocysteine also increases; however, the elevation of homocysteine is seen in a variety of other conditions related to homocysteine and folate metabolism; hence, the most specific and sensitive marker of B12 deficiency currently available is the elevation of methylmalonic acid in urine and serum. In practice, however, homocysteine is easily available and can be considered as the most important surrogate marker of Vitamin B-12 deficiency. The characteristic feature of ITS is the rapid and marked response to Vitamin B12 supplementation. Improvement in appetite occurs; the infant becomes less irritable and more playful, and gradually, neurological symptoms abate. The usual dose is 1000 mcg of Vitamin B-12 given intramuscularly for 7–14 days till there are clear signs of recovery. This is followed by dosing that is less frequent, alternate days, and then once a week. Monthly doses may be given to see that the deficiency does not recur and to cover the crucial periods of recovery. Along with Vitamin B12, they should be supplemented with iron, folic acid, Vitamin D, calcium, and other multivitamins as many times deficiencies coexist.[17,19]
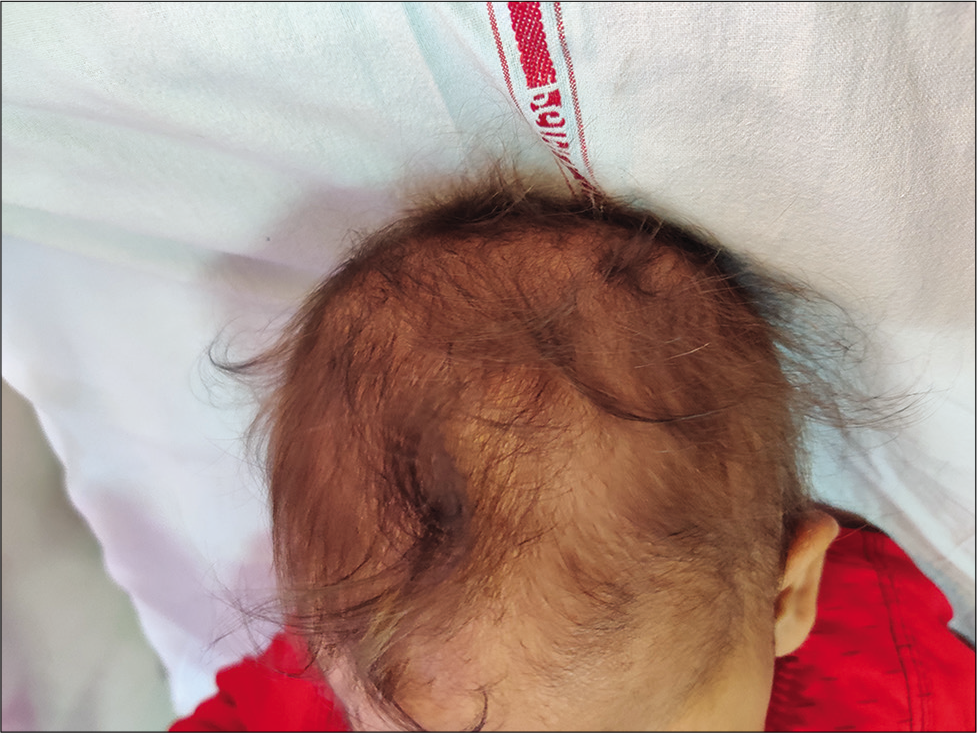
- Sparse hair in a child with nutritional Vitamin B-12 deficiency (infantile tremor syndrome).
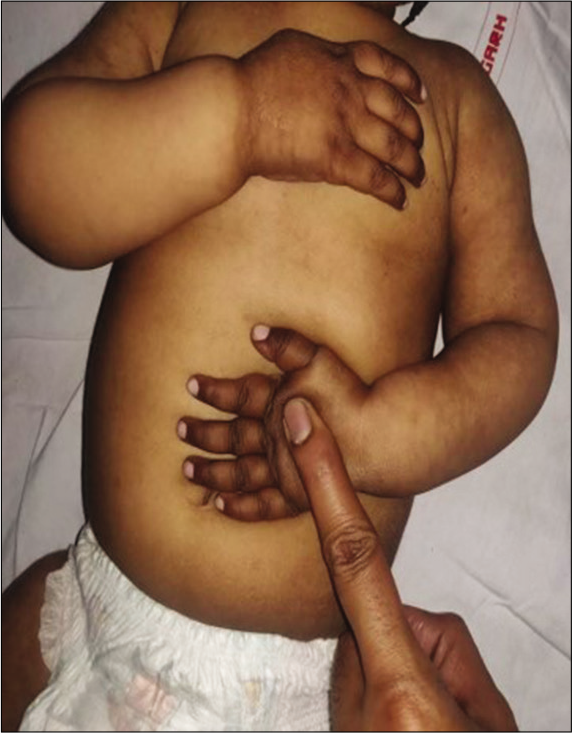
- Hyperpigmentation in a child with infantile tremor syndrome, note the pigmentation on dorsum of hands and reticular pigmentation of arms and forearms.
INHERITED DISORDERS OF INTRACELLULAR COBALAMIN METABOLISM
These are a group of conditions that are all autosomal recessively inherited. They present with a myriad of organ involvement and brain is commonly affected. The disorders either cause isolated homocystinemia, or methylmalonic acidemia or combined homocystinemia and methylmalonic acidemia. Vitamin B-12 is used lifelong to treat these inborn errors of cobalamin pathway with variable benefit.
INHERITED VITAMIN RESPONSIVE DISORDERS PRESENTING WITH ELEVATED HOMOCYSTEINE
Three major disorders in this category include classic homocystinuria due to cystathionine beta-synthase deficiency, combined defects of homocysteine, and methionine and methylenetetrahydrofolate reductase (MTHFR) mutations. All these disorders can present with predominant CNS symptoms such as seizures and mental retardation. Differentiation can be made based on levels of methionine and methylmalonic acid; classic homocystinuria shall have elevated methionine with normal methylmalonic acid levels. Classic homocystinuria responds to pyridoxine, folinic acid, dietary methionine restriction, betaine supplementation, and Cblc defects, and MTHFR deficiency responds to high-dose betaine, hydroxocobalamin, methionine, pyridoxine, and riboflavin.[20]
CEREBRAL FOLATE DEFICIENCY
It is a neurological syndrome associated with a low CSF 5-MTHF in the presence of normal folate metabolism outside the nervous system. This disorder usually presents in infancy with regression, developmental delay, and seizures. The defect is in the impaired folate transporter inside the CNS. This disease responds to oral folinic acid. Secondary forms of cerebral folate deficiency include chronic use of antifolate and anticonvulsant drugs and other conditions such as Rett syndrome, Aicardi-Goutières syndrome, and Kearns-Sayre syndrome.[21]
VITAMIN E
Vitamin E deficiency occurs in chronic malabsorption states such as malabsorption syndromes, cystic fibrosis, and abetalipoproteinemia and manifests with progressive weakness, ataxia, ophthalmoplegia, and loss of position and vibration sense. Treatment consists of usual supplementation with Vitamin E; abetalipoproteinemia usually responds to large doses.
Ataxia with Vitamin E deficiency is usually seen in childhood to early adulthood with clinical features of ataxia, cerebellar signs, areflexia, and neuropathy. Diagnosis is made by the characteristic phenotype along with reduced levels of alpha-tocopherol in serum in the presence of normal levels of lipoproteins and exclusion of malabsorption syndromes. Genetic diagnosis is confirmatory, which shows biallelic pathogenic variations in the TPPA gene.[22]
OTHER VITAMINS
Vitamin C deficiency is now not frequent and is generally seen in malnourished children. It can also occur in children with disabilities or peculiar diets and dietary preferences. Scurvy occurs in severe Vitamin C deficiency and can present with pseudoparalysis. Apart from Vitamin C, Vitamin-K deficiency is also not infrequent despite widespread use of Vitamin-K at birth. Infants present with bleeding manifestations and intracranial hemorrhages can occur [Figure 4]. Infants present with pallor, lethargy, and seizures. Signs of raised ICP like bulging anterior fontanelle are not infrequent.
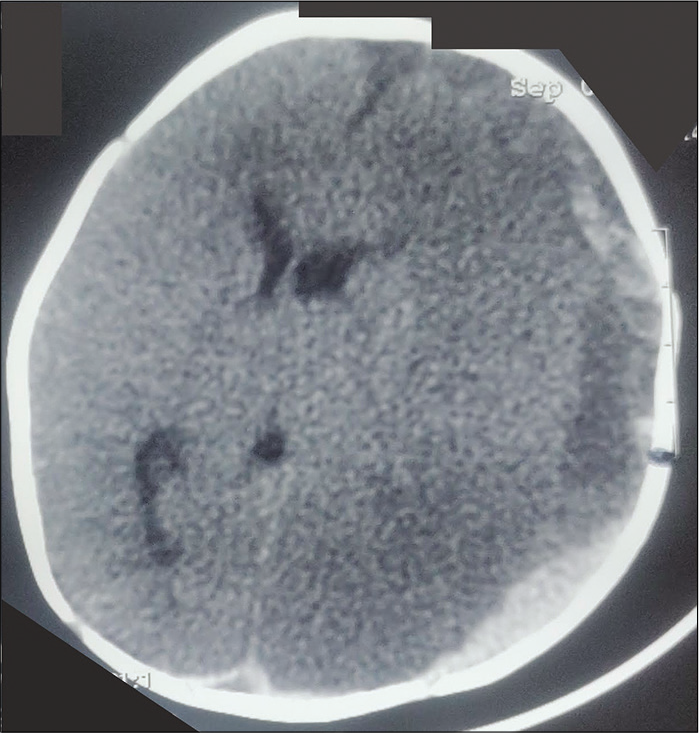
- Plain CT head in an infant with Vitamin K deficiency, large subdural bleed on the left side (black arrow, note the separation of serum and clot giving two density appearances of the bleed), with secondary ischemic changes and midline shift. The baby had presented with pallor, coma, and bulging non-pulsatile anterior fontanelle.
CONCLUSION
For any practicing pediatrician, it is vital to be aware of conditions that are responsive to vitamins. This review presents an overview of some common disorders which are vitamin responsive. However, this list is not exhaustive. It is vital to remain abreast with evolving information of this subject. It is not possible or desirable to remember all such conditions many of which are exceedingly rare. It is crucial that consultation is sought from specialists who have more experience in handling such patients. Many times, the treatment with vitamins is begun empirically awaiting laboratory results. This approach is justified and can prevent and limit disability in affected children.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Vitamin-responsive movement disorders in children. Ann Indian Acad Neurol. 2020;23:325-31.
- [Google Scholar]
- Thiamine-responsive acute severe pulmonary hypertension in exclusively breastfeeding infants: A prospective observational study. Arch Dis Child 2020
- [CrossRef] [PubMed] [Google Scholar]
- Infantile encephalitic beriberi: Magnetic resonance imaging findings. Pediatr Radiol. 2016;46:96-103.
- [CrossRef] [PubMed] [Google Scholar]
- Evaluation of the safety of intravenous thiamine administration in a large academic medical center. J Pharm Pract 2019
- [CrossRef] [PubMed] [Google Scholar]
- Thiamine Deficiency and its Prevention and Control in Major Emergencies, Report No. WHO/NHD/99.13 Geneva: World Health Organization, Department of Nutrition for Health and Development; 1999.
- [Google Scholar]
- Thiamine deficiency, beriberi, and maternal and child health: Why pharmacokinetics matter. Am J Clin Nutr. 2013;98:635-6.
- [CrossRef] [PubMed] [Google Scholar]
- Neurological, psychiatric, and biochemical aspects of thiamine deficiency in children and adults. Front Psychiatry. 2019;10:207.
- [CrossRef] [PubMed] [Google Scholar]
- SLC19A3 gene defects sorting the phenotype and acronyms: Review. Neuropediatrics. 2018;49:83-92.
- [CrossRef] [PubMed] [Google Scholar]
- Riboflavin responsive mitochondrial dysfunction in neurodegenerative diseases. J Clin Med. 2017;6:52.
- [CrossRef] [PubMed] [Google Scholar]
- Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137:44-56.
- [CrossRef] [PubMed] [Google Scholar]
- Current treatment and management of pyridoxine-dependent epilepsy. Curr Treat Options Neurol. 2015;17:335.
- [CrossRef] [PubMed] [Google Scholar]
- Pyridoxine-dependent epilepsy: An expanding clinical spectrum. Pediatr Neurol. 2016;59:6-12.
- [CrossRef] [PubMed] [Google Scholar]
- Biotinidase deficiency: If you have to have an inherited metabolic disease, this is the one to have. Genet Med. 2012;14:565-75.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical practice. Vitamin B12 deficiency. N Engl J Med. 2013;368:149-60.
- [CrossRef] [PubMed] [Google Scholar]
- Developmental outcomes in children with infantile tremor syndrome. Indian J Pediatr. 2020;87:451-3.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile tremor syndrome of the tropics: A neurocutaneous syndrome of infantile B12 deficiency? Eur J Paediatr Neurol. 2017;21:e73.
- [CrossRef] [Google Scholar]
- Reversible basal ganglia changes and metabolic crisis in infantile tremor syndrome. Indian J Pediatr. 2020;87:464-5.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile tremor syndrome: A review and critical appraisal of its etiology. J Pediatr Neurosci. 2016;11:298-304.
- [CrossRef] [PubMed] [Google Scholar]
- What do they have in Common? 2020. Available from: https://www.scielo.br/scielo.php?script=sci_arttext&pid=S2326-45942019000100401 [Last accessed on 2020 Nov 25]
- [Google Scholar]
- Ataxia with Vitamin E deficiency In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, eds. GeneReviews®. Seattle, WA: University of Washington; 1993.
- [Google Scholar]




