
Translate this page into:
Clinical approach to renal tubular acidosis in children
*Corresponding author: Dr. Nivedita Kamath, Department of Pediatric Nephrology, St John’s Medical College Hospital, Bengaluru, Karnataka, India. nkamath25@yahoo.com
-
Received: ,
Accepted: ,
How to cite this article: Reddy S, Kamath N. Clinical approach to renal tubular acidosis in children. Karnataka Paediatr J 2020;35(2):88-94.
Abstract
Renal tubular acidosis (RTA) is a common inherited tubulopathy in children. Proximal RTA, usually secondary to a systemic metabolic disease, is characterized by a generalized dysfunction of the proximal tubule resulting in Fanconi syndrome. Distal RTA occurs due to mutation in the transporters of the distal tubule resulting in acidification defects. Hyperchloremic metabolic acidosis with normal anion gap is the characteristic feature of RTA. In addition to supportive therapy, specific treatment for the underlying etiology and regular monitoring of growth and laboratory parameters are of utmost importance.
Keywords
Renal tubular acidosis
Children
Fanconi syndrome
Distal renal tubular acidosis

INTRODUCTION
The kidneys play a key role in preserving the acid–base homeostasis in the body by excretion of acid and regeneration of bicarbonate. Renal tubular acidosis (RTA) arises from the kidney’s inability to excrete enough acid or retain enough bicarbonate, in the presence of normal renal function, resulting in a clinical syndrome characterized by persistent normal anion gap hyperchloremic metabolic acidosis.[1,2]
ACID–BASE HOMEOSTASIS: ROLE OF THE KIDNEY
Bicarbonate reabsorption in the proximal tubule
The proximal tubule is responsible for reabsorption of 85–90% of filtered bicarbonate through secretion of protons (H+) through the sodium hydrogen exchangers and proton pumps (H+ATPase).[1,3] In the lumen, the secreted hydrogen ions combine with HCO3- to form carbonic acid which rapidly dissociates into carbon dioxide and water, a reaction catalyzed by carbonic anhydrase. The carbon dioxide generated diffuses freely into the proximal tubule cell and reacts with water to form carbonic acid, a reaction catalyzed by carbonic anhydrase (CA II). Bicarbonate ions from the dissociated carbonic acid exit through the basolateral membrane by the sodium bicarbonate exchanger (NBCE1)[4,5] [Figure 1].
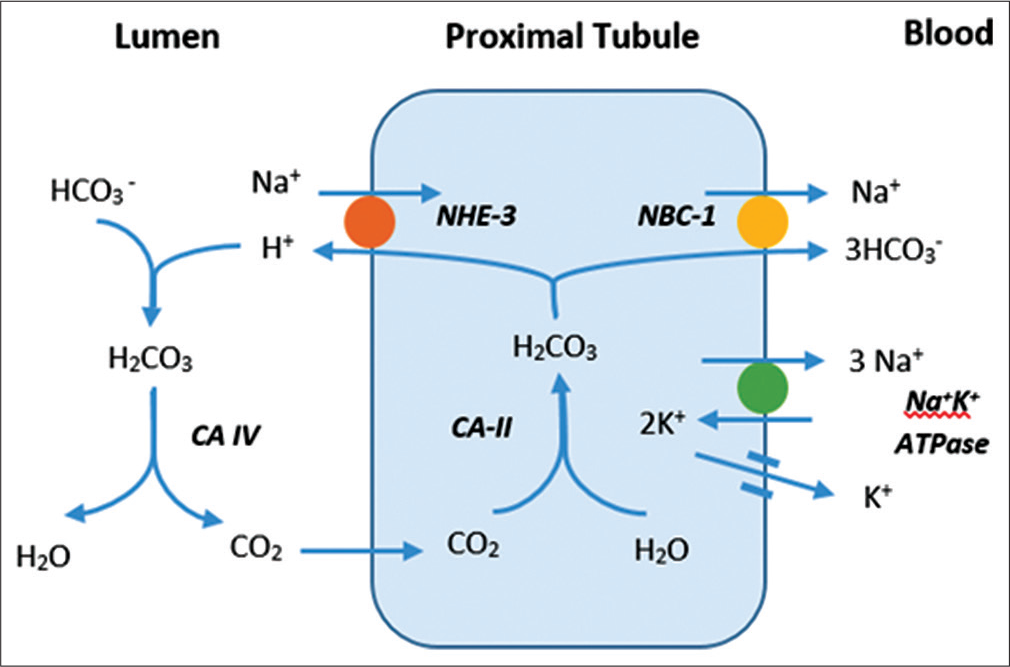
- Bicarbonate reabsorption in proximal tubule.
Urinary acidification in the distal tubule
The acidification of urine occurs in the distal collecting tubule and collecting duct. The intercalated cells A (secrete H+), B (secrete HCO3-) and principal cells (reabsorb sodium, water, and secrete K+) of the distal nephron play a pivotal role in fine tuning acid and base excretion. The generated HCO3- is transported to the blood in exchange for chloride by the basolateral Cl-/HCO3- exchanger. The hydrogen ions secreted into the lumen by intercalated cells A are buffered by titratable acids (phosphate) and ammonia and excreted in the urine[6] [Figure 2].
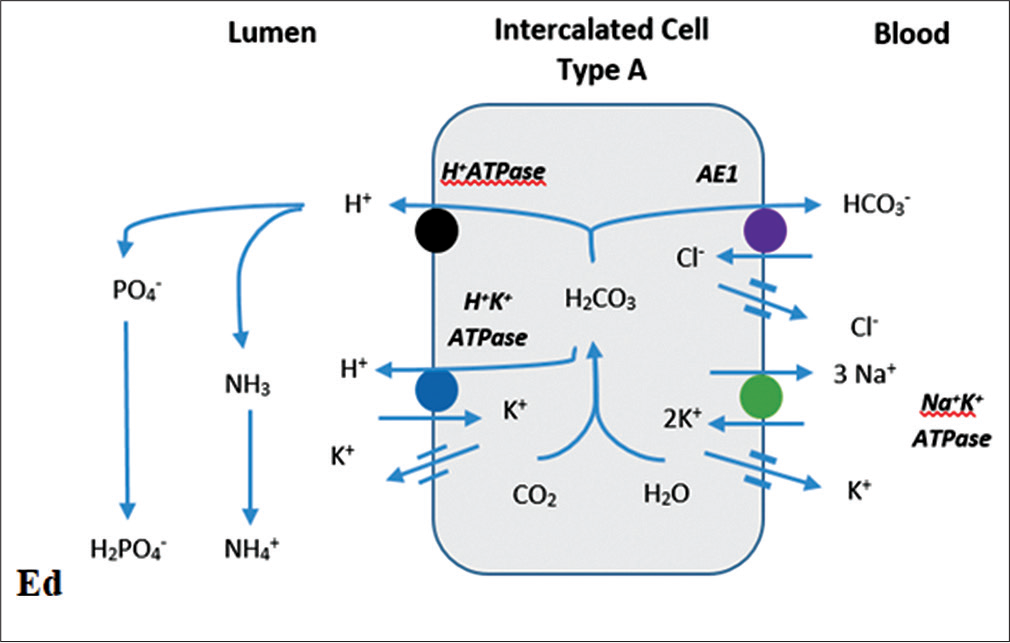
- Distal urinary acidification. NHE 3: Sodium hydrogen exchanger 3, NBC-1: Sodium bicarbonate cotransporter, AE1: Anion exchanger, CA II: Carbonic anhydrase II, CA IV: Carbonic anhydrase IV.
TYPES OF RTA
Type 1 RTA – distal RTA (dRTA)
dRTA, characterized by impaired hydrogen ion secretion in the distal tubules, is commonly seen due to inherited mutations of transporters in the distal tubule.
The hallmark of distal RTA is the inability to lower urine pH maximally in the presence of moderate-to-severe metabolic acidosis. Failure of the distal tubules to excrete the acid load generated by metabolism and in the growing bone in children leads to accumulation of acid and worsening base deficit. Extracellular bicarbonate and hydroxyapatite in the bones serve as buffers to neutralize the accumulated acid. Hypokalemia seen in RTA is due to proximal and distal wasting of sodium, leading to volume contraction and secondary hyperaldosteronism. It is more commonly associated with dRTA as potassium is the only available cation for exchange with sodium and distal secretion of H+ is impaired. Hypercalciuria and hypocitraturia are seen in dRTA which make the children prone for early-onset nephrocalcinosis and nephrolithiasis.[7]
Etiology
-
Inherited distal RTA[8]
Refer to [Table 1].
-
Acquired dRTA
Autoimmune – Sjogren’s syndrome, systemic lupus erythematosus, and Graves’ disease
Medications – Amphotericin, lithium, and aminoglycosides.
| Disorder | Inheritance | Gene | Chromosome | Protein | Extrarenal features |
|---|---|---|---|---|---|
| Distal RTA | |||||
| Autosomal dominant | Autosomal dominant | SLC4A1 | 17q21-q22 | Anion exchanger | Hemolytic anemia |
| Autosomal recessive | Autosomal recessive | ATP6V1B1 ATP6V0A4 |
2q13 7q33-q34 |
H+ATPase | Hearing impairment |
| Proximal RTA | |||||
| Cystinosis | Autosomal recessive | CTNS | 17p13 | Cystinosin | Eyes, CNS, endocrine |
| Dent’s disease Type I Type II |
X linked | CLCN5 OCRL1 (15%) |
Xp11.22 | Chloride channel | Rare |
| Fanconi-Bickel | Autosomal recessive | SLC2A2 | 3q26.1–q26.3 | GLUT2 | Hepatic |
| Galactosemia | Autosomal recessive | GALT | 9p13 | Galactose-1-phosphate uridylyltransferase | Eye, hepatic, CNS |
| Hereditary fructose intolerance | Autosomal recessive | ALDOB | 9q22 | Fructose-1 phosphate aldolase | Hepatic |
| Lowe syndrome | X linked | OCRL1 | Xq26.1 | Phosphatidylinositol 4,5-bisphosphate 5-phosphatase | CNS, eye |
| Tyrosinemia | Autosomal recessive | FAH | 15q23-q25 | Fumarylacetoacetate hydrolase |
Hepatic |
| Wilsons disease | Autosomal recessive | ATP7B | 13q14.3-q21.1 | ATPase copper transporting beta-polypeptide | Hepatic, CNS, eye |
RTA: Renal tubular acidosis, CNS: Central nervous system
Type II RTA – proximal RTA
Proximal RTA is caused by an impairment of bicarbonate reabsorption with intact distal acidification mechanisms and is characterized by decreased renal bicarbonate threshold (14–18 mEq/L), defect in ammonia generation, and ability to lower urine pH <5.5. Hypokalemia is commonly seen in proximal RTA.[9]
Etiology
Proximal RTA usually occurs in association with other tubular defects as a part of the generalized proximal tubular defect – Fanconi syndrome (inherited or acquired). Isolated proximal RTA is rare.
-
Isolated proximal RTA
-
Genetic
Autosomal dominant
Autosomal recessive – with ocular abnormalities (NBCE1), CA II mutation.
Inherited – CA II inhibitors
-
-
Fanconi syndrome[10]
Fanconi syndrome is a generalized dysfunction of the proximal tubule resulting in hypokalemia, polyuria, bicarbonate wasting, glycosuria, low-molecular-weight proteinuria, generalized aminoaciduria, and phosphaturia resulting in hypophosphatemia.
-
Hereditary
Refer to [Table 1].
-
Acquired causes
Autoimmune conditions – Sjogren’s syndrome
Malignancy – Acute lymphatic leukemia, multiple myeloma
Medications – Valproic acid, acetazolamide, cisplatin, lamivudine, tenofovir, outdated tetracyclines, aminoglycosides, Chinese herbs, heavy metals (lead, cadmium, mercury, and copper).
-
Type III RTA – mixed RTA
Type III RTA, an outdated terminology, has combined features of proximal and distal RTA. Association of Type III RTA with osteopetrosis has been commonly described with loss of function mutation of CA II.[11]
Type IV RTA – hyperkalemic RTA
In hyperkalemic RTA, the primary defect lies in the regeneration of bicarbonate secondary to lack of adequate urinary ammonia. This is seen in conditions with aldosterone deficiency or resistance.[12] The acquired causes of aldosterone resistance are the most common causes encountered in clinical practice.
Etiology
-
Hereditary
Congenital hypoaldosteronism
Pseudohypoaldosteronism.
-
Acquired
Urinary tract – posterior urethral valve, reflux nephropathy, pyelonephritis
Autoimmune conditions – interstitial nephritis
Drugs – trimethoprim, nonsteroidal anti-inflammatory drugs, calcineurin inhibitors, angiotensin-converting enzyme inhibitors
CLINICAL CLUES TO SUSPECT RTA IN A CHILD
RTA must be suspected in any child who presents with
Failure to thrive
Resistant rickets
Polyuria, polydipsia
Salt craving – preference for salty foods
Symptoms of hypokalemia
Acidotic breathing
Nephrocalcinosis/nephrolithiasis.
In younger children, polyuria may manifest as recurrent episodes of dehydration in the absence of diarrheal losses, normal urine output in the presence of dehydration, or preference for water over other liquids. Hypokalemia may manifest as abdominal distension, muscle weakness – head lag, or a sudden onset of hypotonia/paralysis usually following an acute illness.
EVALUATION OF RTA[9,13]
The first step in the evaluation of RTA is the identification of hyperchloremic normal anion gap metabolic acidosis in the presence of normal renal function.[9]
-
Anion gap: The anion gap should be calculated by a simultaneous measurement of serum electrolytes and bicarbonate.
Anion gap = [Na+ - (Cl-+ HCO3-) Normal anion gap is 8–12 meq/L.
Anion gap may be affected by the serum albumin levels.
Corrected anion gap = calculated anion gap+ 2.5 (Normal albumin – measured albumin).
The two common conditions causing normal anion gap are diarrhea and RTA.
-
Urine anion gap
The urine anion gap is an indirect measurement of the urinary ammonia excretion in response to metabolic acidosis.
Urinary anion gap = (Na++K+)-Cl- (measured in the urine).
Normally, the urine anion gap is positive. In the presence of metabolic acidosis, the normal kidney is able to generate ammonium (NH4+) and excrete it along with Cl-, making the urine anion gap negative. A persistent positive anion gap in the presence of metabolic acidosis is suggestive of a renal acidification defect.
Caveats: Urine anion gap estimates ammonium excretion only in chronic metabolic acidosis. If the urine pH>6.5, bicarbonate is excreted, hence, urinary anion gap does not reflect urinary ammonium levels.
-
Urine osmolal gap: This is used in situations where the urine anion gap is not reliable.
Urine osmolal gap = measured osmolality – calculated osmolality.
Calculated urine osmolality= 2[Na++K+] + urea + glucose 6 18
Urinary ammonium excretion is estimated to be half the urinary osmolal gap and is considered to be increased if it >100 mosm/kg.
Once, the diagnosis of RTA is confirmed, the next step is to differentiate between the different types of RTA[13]
Urine analysis: A routine analysis of urine for the presence of proteinuria (usually non-nephrotic range, i.e., urine albumin 1+–2+), glycosuria indicates the presence of generalized tubular wasting seen with Fanconi syndrome
-
Urine pH: A urine pH >5.5 in the presence of metabolic acidosis is suggestive of distal acidification defect seen with dRTA. If systemic acidosis is mild, it can be induced using the ammonium chloride loading test. Ammonium chloride is given at 0.1 mg/kg followed by measurement of urine pH every hour for the next 2–8 h. The total plasma CO2 should fall by 3–5 meq/L and urine pH should be <5.5. Ammonium chloride loading test should not be performed in children with moderate-to-severe acidosis (serum bicarbonate <17 meq/L)
Caveat: The urine pH should be measured in a freshly voided sample of urine using a pH meter. The urine pH must always be interpreted with the urine sodium. Low urine sodium will result in low H+ secretion
-
Fractional excretion of bicarbonate: Following a bicarbonate loading test (sodium bicarbonate 0.5 meq/ ml is administered at 3 ml/min), measure the urine pH every 30–60 min till 3 consecutive samples show a pH >7.5.
The fractional excretion of bicarbonate is calculated
The normal fractional excretion of bicarbonate is <5%. A fractional excretion > 15% suggests bicarbonate wasting
-
Urine-blood PCO2: After a load of sodium bicarbonate, the secreted H+ reacts with the luminal HCO3- in the distal lumen forming carbonic acid. In the absence of CA II, this slowly gets converted to CO2 which remains trapped in the lumen. At a urine pH>7.5 and serum bicarbonate 23–25 meq/L, the urine PCO2 should be >70 mmHg. The urine-blood PCO2 > 20 mmHg is seen with normal acid secretion. Urine-blood PCO2<20 mmHg indicates a defect in distal acid secretion
Caveats: Urine for PCO2 should be collected from a freshly voided sample in a sealed syringe using mineral oil and measured by a blood gas analyzer
-
Furosemide fludrocortisone test: The furosemide fludrocortisone test assesses the ability of the distal tubule to secrete acid. Furosemide increases the distal sodium delivery, which, in turn, enhances distal H+ secretion. Fludrocortisone also increases H+ secretion. After an overnight fasting, early morning pH is checked, if urine pH >5.5, oral furosemide is administered at 1 mg/kg and fludrocortisone at 0.025 mg/kg. Urine pH is measured hourly for 4–6 h. A urine pH <5.5 indicates an intact distal acidification mechanism.
Caveat: Ensure normal serum potassium before the test
-
Fractional excretion of phosphate: Fractional excretion of phosphate can be measured using a spot or timed urine sample along with simultaneously measured serum values. The normal fractional excretion of phosphate is 8–15%. Fractional excretion >15% suggests phosphate wasting.
The tubular resorption of phosphate corrected for glomerular filtration rate (TmP/GFR) is a better indicator and can be calculated using the Bijvoet’s nomogram or using the formula. Normal TmP/GFR is 2.8–4.4 mg/dL, which will be reduced in Fanconi syndrome.
Generalized aminoaciduria: Generalized aminoaciduria> 5% is suggestive of proximal tubular wasting
Low-molecular-weight proteinuria: Elevated levels of β2 microglobulinuria is suggestive of Fanconi syndrome
Urinary calcium creatinine ratio: The spot urinary calcium to creatinine ratio is increased (>0.2 in children >2 years of age) in distal RTA and some forms of proximal RTA like Dent’s disease. A 24 h urinary calcium excretion >4 mg/kg/day is considered as hypercalciuria
Hypocitraturia – 24 h urine citrate is estimated using colorimetric methods. 24 h urine citrate levels of <2 mg/kg is termed as hypocitraturia
Ultrasound examination of the kidney – for nephrocalcinosis and nephrolithiasis – seen in distal RTA
Evaluation for extra-renal involvement – Eye examination could reveal cystine crystals in children with cystinosis. Assessment of hearing must be done in all children with dRTA.
The clinical and laboratory presentation of proximal and distal RTA is compared in [Table 2].
| Proximal RTA | Distal RTA | |
|---|---|---|
| Clinical presentation | Usually as a part of a systemic disease; most often metabolic disease | Usually isolated; autosomal recessive forms are associated with hearing loss |
| Bony deformity | Variable | Usually severe |
| Metabolic acidosis | Usually milder, but difficult to correct; requires high doses of bicarbonate supplementation | Severe acidosis; easily corrected with bicarbonate supplementation |
| Serum potassium | Normal/low | Low |
| Urine pH | <5.5 | >5.5 |
| Fractional excretion of bicarbonate | >15% | <5% |
| Urine-blood PCO2 | >20 mmHg | <20 mmHg |
| Phosphaturia and hypophosphatemia | Present (variable) | Absent |
| Tubular defects – low-molecular-weight proteinuria, aminoaciduria, glycosuria | Present (variable) | Absent |
| Hypercalciuria/nephrocalcinosis | Occasionally present | Often present |
RTA: Renal tubular acidosis
TREATMENT FOR RTA
Type I RTA
The goal of treatment is to correct metabolic acidosis, prevent bony deformity, and improve growth. Treatment for dRTA consists of alkali therapy to correct metabolic acidosis. Bicarbonate supplements (4–6 mEq/kg/day in infants to 2–4 mEq/kg/day in children), in the form of potassium citrate formulations, are recommended to ensure normal growth.[14] Hypokalemia improves with correction of acidosis, but some children require long-term supplements (1–2 mEq/kg/day). Potassium citrate in addition to correcting metabolic acidosis and hypokalemia provides citrate which reduces hypercalciuria. It must be remembered that these measures will not retard the progression of nephrocalcinosis or hearing impairment. Regular screening for nephrocalcinosis and hearing impairment must be done and targeted therapy instituted.
Type II RTA
The management of proximal RTA depends on the etiology of tubular dysfunction. The goal of supportive therapy is to promote growth and prevent bony deformities. The main stay of treatment is supplementation of urinary losses of bicarbonate with high doses of alkali (10–15 mEq/kg/day), potassium (1–5 mEq/kg/day), and phosphate (20-40 mg/kg/day). Sodium (3–5 mEq/kg/day) and magnesium (25–50 mg/kg/day) supplements may be needed in some children. Unlike dRTA, children with proximal RTA require high doses of bicarbonate therapy to sustain bicarbonate levels around the renal threshold. Commonly used bicarbonate supplements include:
Syrups – Potrate (Bicarbonate and potassium 2 mEq/ml), Nodosis (0.8 mEq/ml)
Tablets – Sodamint (3.6 mEq/300 mg, 6 mEq/500 mg, 7.8/650 mg), Acidose 500 mg.
Phosphate supplements (Addphos sachet 500 mg, K-Phos tablet 500 mg, and Joules solution 30 mg/ml) are administered at 20–40 mg/kg/day in 3–4 divided doses. The common side effects include diarrhea and abdominal pain. Active Vitamin D supplementation (calcitriol 20–40 ng/kg/day) with regular assessment of urine calcium and annual renal scan to monitor for nephrocalcinosis is recommended.[15] Nutrition in children with RTA is compromised due to polydipsia, polyuria with increased losses of sodium and other nutrients, and poor intake. Intake of calorie dense foods rich in potassium and phosphorus with adequate fluids is encouraged.
Hyperkalemic RTA
The primary treatment is to stabilize serum potassium concentration by stopping all potassium containing medications, restricting dietary sources, and addition of potassium-binding resins (calcium polystyrene sulfonate K-Bind 15 g sachets – 1 g/kg/day in 2–3 divided doses).[12] Bicarbonate supplement should be administered to correct acidosis.
SPECIFIC DISORDERS CAUSING RTA
Proximal RTA
Cystinosis
Cystinosis is the most common hereditary cause of Fanconi syndrome. It is an autosomal recessive lysosomal storage disorder resulting from a defect in the gene CTNS (SLC3A1, SLC7A9) encoding for cystinosin, a lysosomal cystine-proton cotransporter. The commonly affected organs include kidney, eyes, thyroid, pancreas, gonads, and central nervous system. The infantile nephropathic form is seen in 95% of all cases of cystinosis.[16] There is early onset of renal involvement with rapid progression to end-stage renal disease by the end of the first decade. Ocular (corneal deposits in 100% by 18 months), endocrine (50–70% hypothyroidism by the 2nd decade), gonadal (70% have primary hypogonadism), and central nervous system (encephalopathy in 45% by late second decade) involvement are common extrarenal manifestations.[17] Cystine depletion therapy with cysteamine delays end-stage renal disease but does not stop its progression.[18]
Tyrosinemia
Hereditary tyrosinemia Type I is an autosomal recessive inborn error of metabolism, due to deficiency of fumarylacetoacetate hydrolase, mainly affecting the liver and kidney during early infancy. Hepatic manifestations include abnormal synthetic function with coagulopathy, transaminitis, hypoglycemia, acute liver failure in the first few weeks to life, and progression to cirrhosis in early childhood. The spectrum of renal involvement varies in severity from hypophosphatemic rickets, generalized aminoaciduria, and proteinuria with occasional glucosuria (lower incidence in view of decreased plasma glucose levels) to long-term complications of glomerulosclerosis, nephrocalcinosis, and chronic kidney disease. Treatment with low phenylalanine and low tyrosine diet improves renal tubular dysfunction. An effective medical treatment with nitisinone (2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione) has optimal long-term preventive effects if initiated early and when plasma levels are maintained at 40–60 μmol/L.[19]
Dent’s disease
Dent’s disease is an X-linked hereditary form of Fanconi syndrome characterized by proximal tubular dysfunction – low-molecular-weight proteinuria, hypercalciuria, hypophosphatemic rickets, nephrolithiasis, nephrocalcinosis, and progression to end-stage renal disease. More than 50–60% of cases are attributed to mutations in CLCN5 gene (Dent I), 15% have mutations involving the OCRL1 gene (Dent 2). Extrarenal symptoms are rare in Dent’s disease.[20] There is no specific treatment available to retard the progression to end-stage renal disease, which commonly occurs by 30–50 years of age.
Lowe syndrome
Lowe syndrome (oculocerebrorenal syndrome) is an X-linked disorder resulting from mutation in OCRL gene (encoding α-phosphatidylinositol 4, 5-biphosphate phosphatase). Bilateral cataract, generalized hypotonia at birth, and proximal RTA with progression to chronic kidney disease by the second decade are characteristic of Lowe syndrome. Treatment is primarily supportive and involves cataract removal, glaucoma control, targeted rehabilitation therapy, and correction of tubular dysfunction with supplements.[21]
Glycogen storage disorder (GSD)
GSD Type Ia is an autosomal recessive disorder with deficiency of glucose-6-phosphatase-α. Children with GSD I have failure to thrive, hypoglycemia with hepatorenomegaly, hyperuricemia, hyperlipidemia, and proximal RTA.
The other GSD with Fanconi phenotype is Fanconi Bickel syndrome, also referred to as GSD XI, which is caused by a deficiency of GLUT2 transporter, secondary to mutations in SLC2A2 and is characterized by failure to thrive, chronic diarrhea, fasting hypoglycemia, postprandial hyperglycemia, hepatomegaly, and proximal tubular dysfunction typically manifesting in infancy. Dietary modification with restriction of glucose and galactose with incorporation of cornstarch is the main treatment.[22]
Distal RTA
Autosomal dominant
Autosomal dominant distal RTA occurs due to heterozygous mutation of anion exchanger (AE1) in the basolateral surface of the Type A intercalated cells. The usual presentation is during adolescence with features of nephrocalcinosis, nephrolithiasis, occasional rachitic changes, and mild or compensated hyperchloremic metabolic acidosis. Hereditary spherocytosis and ovalocytosis can occur secondary to involvement of AE1 on the erythrocyte membrane.[23,24]
Autosomal recessive
Mutations in ATP6V1B1, ATP6V0A4, and SLC4A1 present in early infancy or childhood with failure to thrive, episodes of vomiting or dehydration, rickets, and nephrocalcinosis with severe metabolic acidosis. These autosomal recessive distal renal tubular disorders are associated with early-onset bilateral sensorineural hearing loss in those with ATP6V1B1 mutations.[25]
Outcome
Proximal RTA secondary to cystinosis, Dent’s disease, etc., have a relentless progression to chronic kidney disease. Although the clinical presentation of proximal tubular dysfunction may be variable, most causes of pRTA are associated with severe growth failure and bony deformities despite adequate treatment. The metabolic abnormalities are often hard to correct despite large supplementary doses.
Distal renal tubular dysfunction is often limited to the kidney alone. They rarely progress to chronic kidney disease. Growth impairment, bony deformities, and metabolic parameters are correctable with adequate therapy.
CONCLUSION
Children with renal tubular acidosis require a thorough evaluation to ascertain the etiology of RTA. Regular long-term follow-up and monitoring of growth, bony deformities and renal function is required.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- New findings on the pathogenesis of distal renal tubular acidosis. Kidney Dis (Basel). 2017;3:98-105.
- [CrossRef] [PubMed] [Google Scholar]
- Integration of acid-base and electrolyte disorders. N Engl J Med. 2014;371:1821-31.
- [CrossRef] [PubMed] [Google Scholar]
- Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol. 2014;9:1627-38.
- [CrossRef] [PubMed] [Google Scholar]
- Core Concepts in the Disorders of Fluid, Electrolytes and Acid-Base Balance. Vol 2. Berlin: Springer; 2013. p. :1-377.
- [CrossRef] [Google Scholar]
- Renal Tubular Acidosis in Children Historical Development of Classification of RTA. 2016
- [CrossRef] [Google Scholar]
- Molecular pathophysiology of renal tubular acidosis. Curr Genomics. 2009;10:51-9.
- [CrossRef] [PubMed] [Google Scholar]
- Evaluation of renal tubular acidosis. Indian J Pediatr. 2007;74:679-86.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol. 2015;30:2099-107.
- [CrossRef] [PubMed] [Google Scholar]
- Pathophysiology of renal tubular acidosis: Core curriculum 2016. Am J Kidney Dis. 2016;68:488-98.
- [CrossRef] [PubMed] [Google Scholar]
- Comprehensive clinical approach to renal tubular acidosis. Clin Exp Nephrol. 2015;19:556-61.
- [CrossRef] [PubMed] [Google Scholar]
- Distal renal tubular acidosis: genetic causes and management. World J Pediatr. 2019;15:422-31.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15:435-55.
- [CrossRef] [PubMed] [Google Scholar]
- The history of cystinosis: Lessons for clinical management. Int J Nephrol. 2011;2011:929456.
- [CrossRef] [PubMed] [Google Scholar]
- Nephropathic cystinosis: Symptoms, treatment, and perspectives of a systemic disease. Front Pediatr. 2018;6:58.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis and treatment of tyrosinemia Type I: A US and Canadian consensus group review and recommendations. Genet Med. 2017;19:1-16.
- [CrossRef] [PubMed] [Google Scholar]
- Late diagnosis of fanconi-bickel syndrome: Challenges with the diagnosis and literature review. J Inborn Errors Metab Screen. 2016;4:1-6.
- [CrossRef] [Google Scholar]
- Distal renal tubular acidosis: A hereditary disease with an inadequate urinary H+ excretion. Nefrologia. 2013;33:289-96.
- [Google Scholar]
- Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol. 2018;31:511-22.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited distal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2178-84.
- [CrossRef] [PubMed] [Google Scholar]




